










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkNova
Print version ISSN 1794-2470
Nova vol.12 no.22 Bogotá July/Dec. 2014
Artículo original producto de la investigación
Análisis Molecular de las Mutaciones 2299delG y C759F en Individuos Colombianos con Retinitis Pigmentosa e Hipoacusia Neurosensorial
Molecular analyses of 2299delG and C759F mutations in Colombian Retinitis Pigmentosa and Sensorineural hearing loss affected individuals
Greizy López1, Nancy Yaneth Gelvez2, Luisa Fernanda Urrego3, Silvia Florez4, David Medina4, Vicente Rodríguez5, Marta Lucía Tamayo2,4
1 Escuela de Ciencias de la Salud, Grupo BIOINNOVA, Universidad Nacional Abierta y a Distancia - UNAD, Colombia.
2 Instituto de Genética Humana, Universidad Javeriana, Bogotá, Colombia.
3 Universidad del Rosario. Bogotá, Colombia.
4 Fundación Oftalmológica Nacional, Bogotá, Colombia.
5 Departamento de ORL, Hospital San Ignacio, Universidad Javeriana, Bogotá, Colombia.
Correspondencia: mtamayo@javeriana.edu.co
RESUMEN
Objetivo: Determinar la presencia de las mutaciones 2299delG y C759F en 37 individuos colombianos no relacionados con asociación de RP e hipoacusia neurosensorial. Materiales y métodos: análisis de secuencia directa del exón 13 del gen USH2A en todos los individuos seleccionados para el estudio. Resultados: la mutación 2299delG fue observada únicamente en individuos con Síndrome de Usher tipo II, mientras que la mutación C759F, no fue observada en ninguno de los individuos del estudio.
Palabras clave: Síndromes de Usher, trastornos sordoceguera, pérdida auditiva sensorineural, retinitis pigmentosa, mutación, genética.
ABSTRACT
Objective: To determine the presence of 2299delG and C759F mutations in 37 non-related subjects from Colombia suffering from RP and sensorineural deafness. Materials and methods: Exon 13 of USH2A gene was directly sequenced in all subjects selected for the study. Results: In this work, the 2299delG mutation was only observed in subjects suffering from Usher syndrome type II while the C759F mutation was not detected in any subject.
Key words: Usher Syndromes, Deaf-Blind Disorders, Sensorineural Hearing Loss, Retinitis Pigmentosa, Mutation, Genetics.
INTRODUCCIÓN
El Síndrome de Usher (USH) es una entidad de herencia autosómica recesiva, que se presenta con hipoacusia neurosensorial, retinitis pigmentosa progresiva y en algunos casos, disfunción vestibular. El Síndrome es la causa más frecuente de sordo-ceguera en el mundo, y constituye el 6% de la población congénitamente sorda y el 18% de toda la población con RP (1); la prevalencia del USH está en un rango de 3.5 a 6.2 casos de cada 100,000 habitantes (2); su frecuencia en Estados Unidos es alrededor de 5/100,000 (1); en Escandinavia de 3.0/100,000 (3) y en Colombia, de 3,2/100.000 habitantes y constituye el 9,6% de la población sorda y el 10% de la población ciega(4-6). Clínicamente, el USH se divide en tres tipos: el tipo I (USH1) se caracteriza por sordera profunda congénita, ausencia de respuesta vestibular y aparición de RP progresiva en la primera o principios de la segunda década de vida; el tipo II (USH2) por sordera congénita de moderada a severa, respuesta vestibular normal, con inicio de RP progresiva en la segunda década de vida; y, el tipo III (USH3), por pérdida auditiva progresiva, respuesta vestibular variable y aparición de RP en edad variable (7).
El USH presenta una significativa heterogeneidad genética: a cada uno de los tres tipos clínicos del síndrome le corresponde uno o más subtipos genéticos. Hasta el momento, se han identificado 14 loci asociados con el USH: seis responsables del fenotipo USH1 (USH1B-K), cuatro del USH2 (USH2A-D) y uno del fenotipo USH3 (USH3A). De ellos, han sido identificados nueve genes responsables: MYO7A para USH1B, USH1C para USH1C, CDH23 para USH1D, PCDH15 para USH1F, SANS para USH1G, y CIB2 para USH1J (8-13); USH2A para USH2A, VLGR1b para USH2C, WHRN para USH2D y USH3A para USH3 (14-18). De todos los anteriormente mencionados, el primer gen identificado fue el USH2A (19), el cual codifica para la proteína Usherina (20, 21).
USH2 es la forma más común de Síndrome de Usher, ya que cuenta para casi la mitad de los casos reportados (22-24). Algunos estudios en población europea y americana han reportado también que del 74 al 83% de los casos USH2, corresponden al subtipo USH2A (16, 25). Inicialmente se describió que el gen USH2A constaba de 21 exones, pero posteriormente se definió que existe una segunda isoforma que comprende 72 exones adicionales a los ya conocidos (26).
Un gran número de mutaciones han sido identificadas en el gen USH2A, pero la más prevalente es la 2299delG (14, 16, 27-31). Esta mutación ha sido identificada en el 25% de la población española con USH2 (27) y es responsable tanto del fenotipo de USH tipo II, como USH atípico (32). Existe evidencia que indica que esta mutación tuvo su origen en la población del sur de Europa (33). Otro cambio frecuente en el gen USH2A, es la mutación C759F, la cual es responsable del fenotipo USH2 (28, 34) y RP sin sordera (35). Ambas mutaciones están localizadas en el exón 13, una región que representa sólo el 6.4% de la región codificante de este gen (36). Dada la prevalencia de estas dos mutaciones y la variabilidad fenotípica que presentan, el objetivo de este trabajo fue identificar estos cambios en individuos colombianos con asociación de RP e hipoacusia neurosensorial congénita.
MATERIALES Y MÉTODOS
Población objeto de estudio
La población objeto de estudio consta de 37 individuos con RP e hipoacusia neurosensorial, que fueron seleccionados en tamizajes realizados por el Instituto de Genética Humana de la Universidad Javeriana y la Fundación Oftalmológica Nacional (FUNDONAL) en 11 ciudades del país, en institutos para ciegos y sordos. Para ello se realizaron exámenes de fondo de ojo y audiometría. A los individuos seleccionados se les tomaron muestras de 7 ml de sangre periférica por punción venosa, previa firma del consentimiento informado. El diagnóstico clínico se realizó con base en la historia clínica completa de los individuos afectados, la evaluación del mayor número posible de familiares, la elaboración del árbol genealógico y exámenes diagnósticos de electrorretinograma (ERG), campimetría, angiografía, fotos de fondo de ojo y audiometría. Este estudio fue aprobado por el Comité de ética de la Pontificia Universidad Javeriana.
Pruebas moleculares
El ADN de las muestras fue extraído por medio de la técnica fenol-cloroformo. Con posterioridad se realizó el análisis de secuenciación del exón 13 del gen USH2A en todos los individuos seleccionados para el estudio. El procedimiento utilizado para la secuenciación fue el siguiente: una vez obtenido el producto de PCR (Polymerase Chain Reaction), se purificó y se realizó la reacción de secuenciación con 2 µl de mezcla de reacción, 2 µl de solución tampón 5X, 1 µl de iniciador y 5 µl de producto purificado. Las condiciones de temperatura fueron: 95 °C por 10 segundos, 50 °C por 5 segundos y 60 °C por 4 minutos, durante 35 ciclos. Se realizó la precipitación de la reacción de secuenciación por el método de etanol. Las análisis de secuenciación se realizaron en el ABI-PRISM 3100-AvantTM con el estuche de secuenciación BigDye terminatorTM, versión 3.1. Finalmente, se analizaron las secuencias con el software DNAstarTM. La secuencia consenso utilizada para el gen USH2A se obtuvo de:
RESULTADOS
Para el estudio fueron seleccionados en total 37 individuos no relacionados distribuidos en tres grupos de la siguiente forma: 1) 26 individuos no relacionados con síndrome de Usher tipo II, 2) cuatro individuos no relacionados con fenotipos similares a Síndrome de Usher pero con un mecanismo de herencia autosómico dominante o presencia de individuos con RP sin sordera e individuos con sordera sin RP dentro de la misma familia, y 3) siete individuos no relacionados con RP e hipoacusia leve no diagnosticados como Síndrome de Usher. Se realizó el análisis de secuenciación automática del exón 13 del gen USH2A en las muestras de los 37 individuos seleccionados.
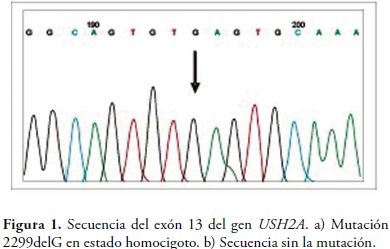
Los resultados del análisis de secuenciación se observan en la Tabla 1. En total, el 18.9% de la población estudiada, porta la mutación 2299delG, ya sea en estado homo o heterocigoto (Figura 1) y ninguno porta la mutación C759F. De la población con diagnóstico de Síndrome de Usher tipo II, cerca del 27% de los propósitos porta la mutación 2299delG, ya sea en estado homocigoto o heterocigoto.
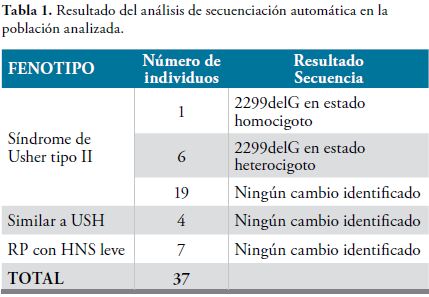
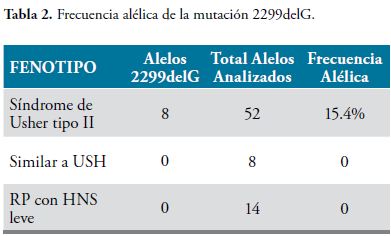
El 3.8% de los individuos con Síndrome de Usher tipo II, portan la mutación 2299delG en estado homocigoto, mientras que el 23.1% la porta en estado heterocigoto. La frecuencia alélica de esta mutación en la población estudiada es de 10.8%, mientras que en la población con Síndrome de Usher tipo II, es de 15.4% (Tabla 2).
El Síndrome de Usher (USH) es una entidad de herencia autosómica recesiva, que se presenta con hipoacusia neurosensorial, retinitis pigmentosa progresiva y en algunos casos, disfunción vestibular. El Síndrome es la causa más frecuente de sordo-ceguera en el mundo, y constituye el 6% de la población congénitamente sorda y el 18% de toda la población con RP (1); la prevalencia del USH está en un rango de 3.5 a 6.2 casos de cada 100,000 habitantes (2); su frecuencia en Estados Unidos es alrededor de 5/100,000 (1); en Escandinavia de 3.0/100,000 (3) y en Colombia, de 3,2/100.000 habitantes y constituye el 9,6% de la población sorda y el 10% de la población ciega(4-6). Clínicamente, el USH se divide en tres tipos: el tipo I (USH1) se caracteriza por sordera profunda congénita, ausencia de respuesta vestibular y aparición de RP progresiva en la primera o principios de la segunda década de vida; el tipo II (USH2) por sordera congénita de moderada a severa, respuesta vestibular normal, con inicio de RP progresiva en la segunda década de vida; y, el tipo III (USH3), por pérdida auditiva progresiva, respuesta vestibular variable y aparición de RP en edad variable (7).
El USH presenta una significativa heterogeneidad genética: a cada uno de los tres tipos clínicos del síndrome le corresponde uno o más subtipos genéticos. Hasta el momento, se han identificado 14 loci asociados con el USH: seis responsables del fenotipo USH1 (USH1B-K), cuatro del USH2 (USH2A-D) y uno del fenotipo USH3 (USH3A). De ellos, han sido identificados nueve genes responsables: MYO7A para USH1B, USH1C para USH1C, CDH23 para USH1D, PCDH15 para USH1F, SANS para USH1G, y CIB2 para USH1J (8-13); USH2A para USH2A, VLGR1b para USH2C, WHRN para USH2D y USH3A para USH3 (14-18). De todos los anteriormente mencionados, el primer gen identificado fue el USH2A (19), el cual codifica para la proteína Usherina (20, 21).
USH2 es la forma más común de Síndrome de Usher, ya que cuenta para casi la mitad de los casos reportados (22-24). Algunos estudios en población europea y americana han reportado también que del 74 al 83% de los casos USH2, corresponden al subtipo USH2A (16, 25). Inicialmente se describió que el gen USH2A constaba de 21 exones, pero posteriormente se definió que existe una segunda isoforma que comprende 72 exones adicionales a los ya conocidos (26).
Un gran número de mutaciones han sido identificadas en el gen USH2A, pero la más prevalente es la 2299delG (14, 16, 27-31). Esta mutación ha sido identificada en el 25% de la población española con USH2 (27) y es responsable tanto del fenotipo de USH tipo II, como USH atípico (32). Existe evidencia que indica que esta mutación tuvo su origen en la población del sur de Europa (33). Otro cambio frecuente en el gen USH2A, es la mutación C759F, la cual es responsable del fenotipo USH2 (28, 34) y RP sin sordera (35). Ambas mutaciones están localizadas en el exón 13, una región que representa sólo el 6.4% de la región codificante de este gen (36). Dada la prevalencia de estas dos mutaciones y la variabilidad fenotípica que presentan, el objetivo de este trabajo fue identificar estos cambios en individuos colombianos con asociación de RP e hipoacusia neurosensorial congénita.
DISCUSIÓN
La frecuencia del USH en Colombia ha sido estimada en 3,2/100.000 habitantes y constituye el 9,6% de la población sorda y el 10% de la población ciega (4-6). Entre la población afectada con USH, casi el 40% corresponde al tipo II de la enfermedad (datos de los autores), y la necesidad de diagnosticar molecularmente estos individuos, ha incrementado el interés hacia la realización de pruebas confiables para detectar las mutaciones propias de la población colombiana, lo cual será de vital importancia en el momento de realizar un diagnóstico temprano e iniciar un proceso de rehabilitación.
La mutación 2299delG es la mutación más frecuente en individuos con USH2 en el mundo y su frecuencia está estimada en un 25% en la población española (27). En el presente estudio, se identificó la mutación 2299deG en un 18.9% de la población analizada y en un 27% de los individuos diagnosticados con USH2. Los individuos con la mutación 2299delG tanto en estado homocigoto como heterocigoto, manifestaron un fenotipo muy similar y homogéneo de USH típico. En cuanto a los individuos que portan la mutación en estado heterocigoto, muy seguramente se trata de heterocigotos compuestos, en quienes hasta el momento no se ha detectado la otra mutación causante. Se observaron algunos casos en los que la hipoacusia se manifestó de forma más severa que en otros, lo cual puede ser causado por la presencia de una segunda mutación más agresiva en algunos de los heterocigotos compuestos. Hasta el momento, no se ha identificado esta mutación en pacientes con RP e hipoacusia leve, ni en pacientes con fenotipo similar a USH en Colombia.
La mutación C759F, consiste en una transversión de una Guanina por una Timina que conduce a un cambio de una Cisteína por una Fenilalanina en el residuo 759, y constituye otro cambio frecuente en el gen USH2A en España (36, 37). En este estudio, la mutación C759F no fue identificada en ninguno de los individuos de la población analizada, contrario a lo que se esperaba, lo que indica que la población colombiana no puede, en todos los casos, ser comparable con la población española. Estos resultados sugieren que la población colombiana podría tener mutaciones propias; debido, tal vez, a diferentes rutas de migración, el gran bagaje indígena o incluso, mutaciones de tipo efecto fundador. Este mismo fenómeno se ha reportado en otras poblaciones, como es el caso de Japón, en donde existe una marcada diferencia entre su espectro mutacional y el de otras poblaciones (38, 39). La población colombiana podría estar mostrando un comportamiento similar al de otras poblaciones como la china o la finlandesa (40, 41).
Se conoce que mutaciones en el gen USH2A son responsables de un amplio espectro de fenotipos y, en especial, las mutaciones 2299delG y C759F, que se han visto asociadas a fenotipos tanto de Síndrome de Usher tipo II, Síndrome de Usher atípico y RP aislada. En los reportes se ha descrito como Síndrome de Usher atípico aquellos individuos que presentan RP e hipoacusia neurosensorial de herencia autosómica recesiva, pero similar al fenotipo del USH3, es decir, con progresión de la hipoacusia (32, 36). En el presente estudio, los individuos reportados con un fenotipo similar a USH, son individuos que si se analizan de forma particular, podrían ser diagnosticados como USH tipo II, pero que al analizar el conjunto familiar, se hace evidente un patrón de herencia autosómico dominante que no coincide con los parámetros de diagnóstico del síndrome. En otras de las familias, se observan individuos con lo que parecería ser USH, pero con hermanos con RP aislada y otros con hipoacusia aislada; por lo tanto, no se puede definir en ellos el diagnóstico clínico de USH atípico hasta que se logre identificar los genes responsables.
En conclusión, este estudio muestra que el 27% de la población colombiana con diagnóstico clínico de Síndrome de Usher tipo II porta la mutación 2299delG, y esta frecuencia es muy similar a la reportada en España, que es del 25% (27). La mutación C759F, no ha sido identificada hasta el momento en individuos con Síndrome de Usher tipo II. Para el futuro, sería importante realizar el mismo tamizaje mutacional en individuos con RP aislada, para poder establecer la frecuencia de las mutaciones en esta población.
AGRADECIMIENTOS
Esta investigación fue financiada por COLCIENCIAS con el proyecto “LIGAMIENTO GENÉTICO Y ESTUDIOS MUTACIONALES EN 10 FAMILIAS COLOMBIANAS CON SíNDROME DE USHER”, código no. 1203-04-11732, contrato no. 141-2002 y por el Instituto de Genética Humana de la Universidad Javeriana en Bogotá. Proyecto No 1045 titulado "Ligamiento génico y estudios mutacionales en 10 familias colombianas con síndrome de Usher”. Agradecemos al Hospital Universitario San Ignacio por los exámenes audiométricos; a la Fundación Oftalmológica Nacional y a la Clínica Horus por los exámenes oftalmológicos. Al Boys Town Nacional Research Hospital en Omaha, NE, USA. Un especial reconocimiento a los pacientes y sus familiares que gentilmente aceptaron participar en este estudio, así como a todos los institutos de y para niños ciegos y sordos del país.
REFERENCIAS
1. Boughman JA, Vernon M, Shaver KA. Usher syndrome: definition and estimate of prevalence from two high-risk populations. J Chronic Dis. 1983;36(8):595-603. [ Links ]
2. Keats BJ, Corey DP. The usher syndromes. Am J Med Genet. 1999 Sep 24;89(3):158-66. [ Links ]
3. Hallgren B. Retinitis pigmentosa combined with congenital deafness; with vestibulo-cerebellar ataxia and mental abnormality in a proportion of cases: A clinical and genetico-statistical study. Acta Psychiatr Scand Suppl. 1959;34(138):1-101. [ Links ]
4. Tamayo ML, Bernal JE, Tamayo GE, Frias JL, Alvira G, Vergara O, et al. Usher syndrome: results of a screening program in Colombia. Clin Genet. 1991 Oct;40(4):304-11. [ Links ]
5. Tamayo ML, Bernal JE, Tamayo GE, Frias JL. Study of the etiology of deafness in an institutionalized population in Colombia. Am J Med Genet. 1992 Nov 1;44(4):405-8. [ Links ]
6. Tamayo ML, Maldonado C, Plaza SL, Alvira GM, Tamayo GE, Zambrano M, et al. Neuroradiology and clinical aspects of Usher syndrome. Clin Genet. 1996 Sep;50(3):126-32. [ Links ]
7. Pakarinen L, Tuppurainen K, Laippala P, Mantyjarvi M, Puhakka H. The ophthalmological course of Usher syndrome type III. Int Ophthalmol. 1995;19(5):307-11. [ Links ]
8. Kimberling WJ, Moller CG, Davenport S, Priluck IA, Beighton PH, Greenberg J, et al. Linkage of Usher syndrome type I gene (USH1B) to the long arm of chromosome 11. Genomics. 1992 Dec;14(4):988-94. [ Links ]
9. Smith RJ, Lee EC, Kimberling WJ, Daiger SP, Pelias MZ, Keats BJ, et al. Localization of two genes for Usher syndrome type I to chromosome 11. Genomics. 1992 Dec;14(4):995-1002. [ Links ]
10. Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001 Jan;68(1):26-37. [ Links ]
11. Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, et al. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet. 2001 Jul;69(1):25-34. [ Links ]
12. Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet. 2003 Mar 1;12(5):463-71. [ Links ]
13. Riazuddin S, Belyantseva IA, Giese AP, Lee K, Indzhykulian AA, Nandamuri SP, et al. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet. 2012 Nov;44(11):1265-71. [ Links ]
14. Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, et al. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998 Jun 12;280(5370):1753-7. [ Links ]
15. Hmani M, Ghorbel A, Boulila-Elgaied A, Ben Zina Z, Kammoun W, Drira M, et al. A novel locus for Usher syndrome type II, USH2B, maps to chromosome 3 at p23-24.2. Eur J Hum Genet. 1999 Apr;7(3):363-7. [ Links ]
16. Weston MD, Eudy JD, Fujita S, Yao S, Usami S, Cremers C, et al. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am J Hum Genet. 2000 Apr;66(4):1199-210. [ Links ]
17. Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet. 2004 Feb;74(2):357-66. [ Links ]
18. Yang J, Wang L, Song H, Sokolov M. Current understanding of usher syndrome type II. Front Biosci (Landmark Ed). 2012; 17:1165-83. [ Links ]
19. Kimberling WJ, Weston MD, Moller C, Davenport SL, Shugart YY, Priluck IA, et al. Localization of Usher syndrome type II to chromosome 1q. Genomics. 1990 Jun;7(2):245-9. [ Links ]
20. Joensuu T, Hamalainen R, Yuan B, Johnson C, Tegelberg S, Gasparini P, et al. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet. 2001 Oct;69(4):673-84. [ Links ]
21. Adato A, Vreugde S, Joensuu T, Avidan N, Hamalainen R, Belenkiy O, et al. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur J Hum Genet. 2002 Jun;10(6):339-50. [ Links ]
22. Hope CI, Bundey S, Proops D, Fielder AR. Usher syndrome in the city of Birmingham--prevalence and clinical classification. Br J Ophthalmol. 1997 Jan;81(1):46-53. [ Links ]
23. Rosenberg T, Haim M, Hauch AM, Parving A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin Genet. 1997 May;51(5):314-21. [ Links ]
24. Ouyang XM, Hejtmancik JF, Jacobson SG, Li AR, Du LL, Angeli S, et al. Mutational spectrum in Usher syndrome type II. Clin Genet. 2004 Apr;65(4):288-93. [ Links ]
25. Pieke-Dahl S, van Aarem A, Dobin A, Cremers CW, Kimberling WJ. Genetic heterogeneity of Usher syndrome type II in a Dutch population. J Med Genet. 1996 Sep;33(9):753-7. [ Links ]
26. van Wijk E, Pennings RJ, te Brinke H, Claassen A, Yntema HG, Hoefsloot LH, et al. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am J Hum Genet. 2004 Apr;74(4):738-44. [ Links ]
27. Beneyto MM, Cuevas JM, Millan JM, Espinos C, Mateu E, Gonzalez-Cabo P, et al. Prevalence of 2314delG mutation in Spanish patients with Usher syndrome type II (USH2). Ophthalmic Genet. 2000 Jun;21(2):123-8. [ Links ]
28. Dreyer B, Tranebjaerg L, Rosenberg T, Weston MD, Kimberling WJ, Nilssen O. Identification of novel USH2A mutations: implications for the structure of USH2A protein. Eur J Hum Genet. 2000 Jul;8(7):500-6. [ Links ]
29. Leroy BP, Aragon-Martin JA, Weston MD, Bessant DA, Willis C, Webster AR, et al. Spectrum of mutations in USH2A in British patients with Usher syndrome type II. Exp Eye Res. 2001 May;72(5):503-9. [ Links ]
30. Bernal S, Ayuso C, Antinolo G, Gimenez A, Borrego S, Trujillo MJ, et al. Mutations in USH2A in Spanish patients with autosomal recessive retinitis pigmentosa: high prevalence and phenotypic variation. J Med Genet. 2003 Jan;40(1):e8. [ Links ]
31. Lopez G, Gelvez NY, Tamayo M. [Mutational frequencies in usherin(USH2A gene) in 26 Colombian individuals with Usher syndrome type II]. Biomedica. 2011 Mar;31(1):82-90. [ Links ]
32. Liu XZ, Hope C, Liang CY, Zou JM, Xu LR, Cole T, et al. A mutation (2314delG) in the Usher syndrome type IIA gene: high prevalence and phenotypic variation. Am J Hum Genet. 1999 Apr;64(4):1221-5. [ Links ]
33. Aller E, Larrieu L, Jaijo T, Baux D, Espinos C, Gonzalez-Candelas F, et al. The USH2A c.2299delG mutation: dating its common origin in a Southern European population. Eur J Hum Genet. 2010 Jul;18(7):788-93. [ Links ]
34. Najera C, Beneyto M, Blanca J, Aller E, Fontcuberta A, Millan JM, et al. Mutations in myosin VIIA (MYO7A) and usherin (USH2A) in Spanish patients with Usher syndrome types I and II, respectively. Hum Mutat. 2002 Jul;20(1):76-7. [ Links ]
35. Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000 Jun;66(6):1975-8. [ Links ]
36. Aller E, Najera C, Millan JM, Oltra JS, Perez-Garrigues H, Vilela C, et al. Genetic analysis of 2299delG and C759F mutations (USH2A) in patients with visual and/or auditory impairments. Eur J Hum Genet. 2004 May;12(5):407-10. [ Links ]
37. Garcia-Garcia G, Aparisi MJ, Jaijo T, Rodrigo R, Leon AM, Avila-Fernandez A, et al. Mutational screening of the USH2A gene in Spanish USH patients reveals 23 novel pathogenic mutations. Orphanet J Rare Dis. 2011;6:65. [ Links ]
38. Nakanishi H, Ohtsubo M, Iwasaki S, Hotta Y, Usami S, Mizuta K, et al. Novel USH2A mutations in Japanese Usher syndrome type 2 patients: marked differences in the mutation spectrum between the Japanese and other populations. J Hum Genet. 2011 Jul;56(7):484-90. [ Links ]
39. Zhao Y, Hosono K, Suto K, Ishigami C, Arai Y, Hikoya A, et al. The first USH2A mutation analysis of Japanese autosomal recessive retinitis pigmentosa patients: a totally different mutation profile with the lack of frequent mutations found in Caucasian patients. J Hum Genet. 2014 Sep;59(9):521-8. [ Links ]
40. Vastinsalo H, Jalkanen R, Bergmann C, Neuhaus C, Kleemola L, Jauhola L, et al. Extended mutation spectrum of Usher syndrome in Finland. Acta Ophthalmol. 2013 Jun;91(4):325-34. [ Links ]
41. Huang XF, Xiang P, Chen J, Xing DJ, Huang N, Min Q, et al. Targeted exome sequencing identified novel USH2A mutations in Usher syndrome families. PLoS One. 2013;8(5):e63832. [ Links ]
