










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIngeniería y Ciencia
Print version ISSN 1794-9165
ing.cienc. vol.7 no.13 Medellín Jan./June 2011
Estudio teórico de las propiedades elásticas de los minerales Cu3TMSe4 (TM = V, Nb, Ta) por medio de cálculos atomísticos de primeros-principios
Estudo teorico das propriedades elasticas dos minerales Cu3TMSe4 (TM = V, Nb, Ta) por médio de cálculos atomisticos de primeiros princípios
Theoretical study of the elastic properties of the minerals Cu3TMSe4 (TM = V, Nb, Ta) by means of atomistic first-principles calculations
Carlos M. Ruiz1 y Jorge M. Osorio-Guillén2
1 Físico,cmruiz@santafe-conicet.gov.ar,Estudiante de Doctorado, Universidad Nacional del Litoral, Santa Fe - Argentina.
2 Doctor en Física, jorge.osorio@fisica.udea.edu.co, Profesor-Asistente, Instituto de Física - Universidad de Antioquia, Medellín - Colombia.
(Recepción: 11-agost-2010. Modificación: 24-dic-2010. Aceptación: 07-feb-2011)
Se aceptan comentarios y/o discusiones al artículo
Resumen
Las propiedades elásticas de la familia de los minerales isoestructurales Cu3VSe4, Cu3NbSe4 y Cu3TaSe4 han sido calculadas por primera vez usando el estado del arte en cálculos atomísticos de primeros-principios, utilizando la Teoría de los Funcionales de la Densidad y la Aproximación del Gradiente Generalizado para el funcional de la energía de intercambio-correlación. Las propiedades elásticas calculadas son el módulo volumétrico (B), las constantes elásticas (c11, c12 y c44), el factor de anisotropía de Zener (A), el módulo de cizalladura isotrópico (G), el módulo de Young (Y), y la razón de Poisson (ν). A través de estas cantidades también hemos calculado otras propiedades termodinámicas tales como la velocidad promedio del sonido transversal (st) y longitudinal (sl) y la temperatura de Debye (ΘD). Los valores calculados de B , c11, c12 y c44, G, Y , y ν nos llevan a la conclusión que estos compuestos son compresibles, frágiles y quebradizos.
Palabras claves: propiedades elásticas, constantes elásticas, propiedades mecánicas, teoría de los funcionales de la densidad.
Resumo
As propriedades elásticas da família de minerais isoestruturais Cu3VSe4, Cu3NbSe4 e Cu3TaSe4 foi calculado pela primeira vez usando o estado da arte em cálculos atomísticos de primeiros princípios, utilizandose Teoria do Funcional da Densidade e Aproximação das Gradiente Generalizada para funcional a energia de intercâmbio-correlação. As propriedades elásticas calculados são módulo volume (B), as constantes elásticas (c11, c12 e c44), o fator de anisotropia de Zener (A), o módulo de cisalhamento isotrópico (G), módulo de Young (Y), e Poisson ratio (ν). Através destas quantidades também calculamos outras propriedades termodinâmicas como a velocidade média do som transversal (st) e longitudinal (sl) e temperatura de Debye (ΘD). Os valores calculados de B, c11, c12 e c44, G, Y e ν nos levam a concluir que estes compostos são compressíveis, frágeis e quebradiços.
Palavras chaves: propiedades elásticas, constantes elásticas, propiedades mecânicas, teoria do funcional da densidade.
Abstract
The elastic properties of the family of isostructural minerals Cu3VSe4, Cu3NbSe4and Cu3TaSe4have been calculated for the first time using the state of the art in first-principles atomistic calculations, using Density Functional Theory and the Generalized Gradient Approximation for the exchangecorrelation energy functional. The elastic properties calculated are bulk modulus (B), the elastic constants (c11, c12 and c44), the Zener anisotropy factor (A), the isotropic shear modulus (G), the Young modulus (Y), and the Poisson ratio (ν). By means of these quantities we also computed other thermodynamic properties such as the average transversal (st) and longitudinal (sl) sound velocities and the Debye temperature (ΘD). The calculated values of B, c11, c12 and c44, G, Y and ν lead us to the conclusion that these compounds are compressible, fragile and brittle.
Key words: elastic properties, elastic constants, mechanical properties, density functional theory.
1 Introducción
Los minerales isoestructurales Cu3VSe4, Cu3NbSe4y Cu3TaSe4pertenecen a la famila de las sulvanitas[1, 2, 3, 4, 5, 6, 7], los cuales despertaron gran interés en la comunidad científica debido a que presentan conductividad mixta, i.e., existe conductividad por huecos y conductividad iónica[8]. Estudios termodinámicos teóricos y experimentales sobre la sulvanita atribuyen la conductividad de huecos a la presencia de vacancias de Cu y la conductividad iónica a temperatura ambiente como resultado, presumiblemente, de impurezas intersticiales de iones de Cu+[8]. Esta buena conductividad iónica a temperatura ambiente originó el estudio de inserción-desinserción de iones de Li+ en la sulvanita y su posible utilización tecnológica como electrodos en baterías de Litio recargables[9, 10]. El voltaje de circuito abierto medido en el proceso de descarga muestra varios plateaus, siendo el máximo de 3 V[10]. Sin embargo, estudios in-situ de rayos-X de las estructuras cristalinas en el ciclo de carga muestran que estas no tienen nada que ver con las estructuras cristalinas iniciales, lo cual indica una transformación de fase cristalográfica irreversible[10], sugiriendo que la vida útil de estos electrodos no es muy prometedora para la industria de baterías de Litio recargables. Estudios de las propiedades vibracionales de las sulvanitas por medio de reflectividad en el infrarrojo y dispersión de Raman revelan que los modos normales de vibración en estos compuestos se comportan como en la mayoría de los semiconductores y que algunos otros modos son altamente anarmónicos debido, posiblemente, a desorden configuracional de los iones de Cu[7]. Por otra parte, estudios de los estados de valencia de Cu y V en Cu3VS4por medio de espectroscopía de rayos-X[11, 12] muestran que en este compuesto hay presencia de iones de Cu2+, Cu+, V2+y V5+[13, 14]. Estos resultados llevan a la conclusión de que probablemente los estados de valencia en la sulvanita no están bien definidos (los dos posibles estados formales de oxidación son 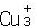 TM5+
TM5+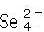 y
y 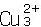 TM2+) y fluctúan con Cu resonando entre los estados cuproso y cúprico y V entre estados 5+ y más bajos. Recientemente se han estudiado las propiedades ópticas y de transporte de películas delgadas de las sulvanitas[15, 16]; las medidas del espectro de reflexión y transmisión han mostrado que Cu3TaS4 es transparente en el visible mientras Cu3TaSe4 es ligeramente coloreado. Los valores del bangap estimados para Cu3TaS4 y Cu3TaSe4son de 2,70 eV y 2,35 eV, respectivamente[16]. Las medidas de transporte de las películas policristalinas sin dopar indican una conductividad cercana a 1 S/cm, con una densidad de huecos en el rango de 5-7×1019cm−3, y una movilidad de 0,2 0,4 cm2/Vs [15].
TM2+) y fluctúan con Cu resonando entre los estados cuproso y cúprico y V entre estados 5+ y más bajos. Recientemente se han estudiado las propiedades ópticas y de transporte de películas delgadas de las sulvanitas[15, 16]; las medidas del espectro de reflexión y transmisión han mostrado que Cu3TaS4 es transparente en el visible mientras Cu3TaSe4 es ligeramente coloreado. Los valores del bangap estimados para Cu3TaS4 y Cu3TaSe4son de 2,70 eV y 2,35 eV, respectivamente[16]. Las medidas de transporte de las películas policristalinas sin dopar indican una conductividad cercana a 1 S/cm, con una densidad de huecos en el rango de 5-7×1019cm−3, y una movilidad de 0,2 0,4 cm2/Vs [15].
Debido a las propiedades físicas descritas anteriormente, estos compuestos podrían llegar a ser buenos candidatos para aplicaciones en dispositivos opto-electrónicos[15, 16, 17, 18, 19, 20], e.g., en capas absorbentes tipop en heteroestructuras para aplicación en celdas solares[15, 16, 21], espejos dieléctricos[16], etc. Otro factor positivo en la posible utilización de las sulvanitas en dispositivos electrónicos es la similitud entre las constantes de red de estos materiales (en especial de Cu3TaSe4) y el Silicio, haciendo posible el crecimiento heteroepitaxial de películas delgadas de Cu3TMSe4sobre sustratos de Si[16]; así como fabricar soluciones sólidas de Cu3TaSe4y Cu
En este trabajo se presenta por primera vez un estudio teórico de las propiedades elásticas de las sulvanitas Cu3TMSe4 (TM = V, Nb, Ta) por medio del estado del arte en cálculos atomísticos de primeros-principios, utilizando la Teoría de los Funcionales de la Densidad y la Aproximación del Gradiente Generalizado para el funcional de la energía de intercambio-correlación. Este estudio además es el primer reporte, hasta donde los autores conocen, experimental y/o teórico, de los valores de las propiedades elásticas de estos compuestos, las cuales son muy importantes en el conocimiento del efecto de las tensiones mecánicas resultantes en el proceso de crecimiento de cristales y de dispositivos semiconductores. Tales efectos mecánicos tienen un gran impacto en el comportamiento y la fiabilidad de estos dispositivos, e.g., hay varios indicios de que las dislocaciones inducidas durante la fabricación de estos dispositivos tienen posteriormente un efecto muy perjudicial en su rendimiento operativo y el fenómeno de degradación[24].
2 Teoría
Las propiedades elásticas de un material son determinadas por las simetrías que éste presente, de modo que entre más simetrías menos constantes elásticas hay que determinar. Así, por ejemplo, en un cristal cúbico que tiene 48
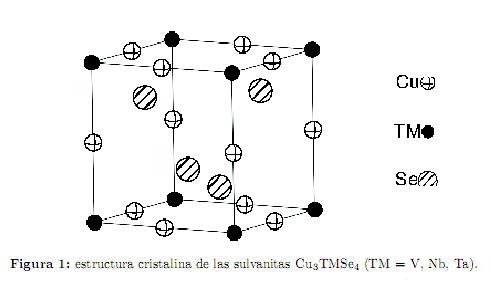
operaciones de simetría, se tienen tres constantes elásticas (c11, c12 y c44); mientras que para un cristal hexagonal, que cuenta con 24 operaciones de simetría, se tienen cinco constantes elásticas (c11, c12, c13, c33 y c55). Los compuestos de la familia isoestructural de las sulvanitas Cu3TMSe4 (TM = V, Nb, Ta) cristalizan en un sistema cúbico simple, perteneciente al grupo espacial 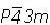 (número 215), con ocho átomos por celda unitaria. Esta estructura esta formada por un arreglo tridimensional de los tetraedros CuSe4 y TMSe4 conectados por sus aristas comunes; además los tetraedros CuSe4 comparten vértices entre ellos. Los iones Cu, TM y Se toman las posiciones de Wyckoff 3d: (1/2,0,0), 1a: (0,0,0) y 4e: (x, x, x), respectivamente[1, 3, 5]. La estructura cristalina de estos materiales se muestra en la figura 1. Para calcular las constantes elásticas de un cuerpo podemos usar las deformaciones que éste experimenta por la acción de fuerzas aplicadas. Para realizar este cálculo definimos el vector de desplazamiento
(número 215), con ocho átomos por celda unitaria. Esta estructura esta formada por un arreglo tridimensional de los tetraedros CuSe4 y TMSe4 conectados por sus aristas comunes; además los tetraedros CuSe4 comparten vértices entre ellos. Los iones Cu, TM y Se toman las posiciones de Wyckoff 3d: (1/2,0,0), 1a: (0,0,0) y 4e: (x, x, x), respectivamente[1, 3, 5]. La estructura cristalina de estos materiales se muestra en la figura 1. Para calcular las constantes elásticas de un cuerpo podemos usar las deformaciones que éste experimenta por la acción de fuerzas aplicadas. Para realizar este cálculo definimos el vector de desplazamiento
u(r) = r′ − r,
donde r es la posición de algún punto en el cuerpo sin deformar y r′ su posición después de la deformación. Si se consideran pequeñas deformaciones, las distancias entre los puntos antes y después de la deformación están dadas por
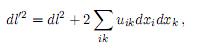
donde el tensor de deformación uik se define como[25]
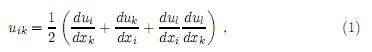
que por ser simétrico (uik = uki) permite elegir en cualquier punto del sólido un sistema de ejes coordenados (ejes principales) de tal manera que solo las componentes diagonales u11, u22 y u33, sean diferentes de cero. En casos donde uiy sus derivadas sean pequeñas, es posible despreciar el último término en (1) y así el tensor de deformación se reduce a
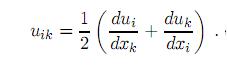
Haciendo uso de la relación entre la energía interna E y el tensor de deformación, y si además expandimos la energía en potencias de uik obtenemos
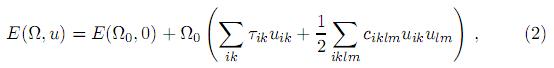
donde E(Ω0, 0) es la energía interna del cristal sin deformar, Ω0 es el volumen del cristal sin deformar y τik son los coeficientes de los términos correspondientes a la presión en una dirección determinada. El tensor de rango cuatro ciklm representa las constantes elásticas, y tiene las mismas propiedades de simetría que el tensor de esfuerzo, ciklm = ckilm = cikml = clmik. La ecuación (2) puede ser simplificada utilizando la notación de Voigt[26], en la cual se escribeciklm como cjn, con j y n tomando valores de 1 a 6 en correspondencia con las componentes cartesianas xx, yy, zz, yz, zx, xy
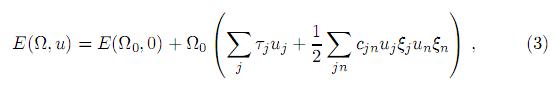
donde el factor ξ toma los valores ξ = 1 si j= 1,2,3 o ξ = 2 si j= 4,5,6. Las constantes elásticas cij son obviamente los coeficientes del término de segundo orden en la expansión de la energía
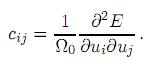
Es claro que el cálculo de las constantes elásticas requiere encontrar la energía interna del cuerpo sin deformar y deformado. El cálculo de todas estas energías se realizó a nivel atomístico, por lo tanto se debe hacer uso de la mecánica cuántica. Nosotros utilizamos la Teoría de los Funcionales de la Densidad[27, 28] y la Aproximación del Gradiente Generalizado[29] para el funcional de la energía de intercambio-correlación. Para resolver las ecuaciones de Kohn-Sham[28] usamos el método del Proyector de Ondas Aumentadas[30] tal como está implementado en el código VASP[31, 32]. Las configuraciones atómicas de referencia son 3p63d104s1 para Cu, 3p63d44s1 para V, 4p64d45s1 para Nb, 5p65d46s1 para Ta, y 4s24p4 para Se; donde solo los electrones que son tratados como electrones de valencia son enumerados explícitamente. La energía de corte en la expansión de ondas planas ha sido convergida hasta que la energía total cambió alrededor de 1 meV/celda unitaria; este valor es de 497,6 eV. La integración en la primera zona de Brillouin se realizó en una malla 6 × 6 × 6 usando el esquema de Monkhorst[33] para la relajación atómica. En el cálculo de la densidad de carga y las energías internas totales utilizamos una malla de 12 × 12 × 12 y el método del tetraedro incluyendo correcciones de Blöchl.
3 Resultados y Discusión
Para sistemas cúbicos se tienen tres constantes elásticas independientes c11, c12 y c44 que pueden ser obtenidas calculando la energía total como función del tensor de deformación de la forma propuesta por Söderlind[34]. Para calcular c11 y c12 se considera la distorsión tetragonal que preserva el volumen (figura 2)
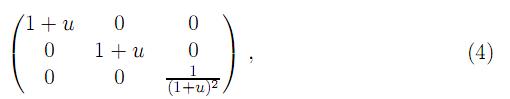
donde u es el valor del parámetro de distorción. La energía por celda unitaria en función de u, asociada con este esfuerzo, tiene la forma

donde 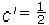 (c11 − c12). De modo que al calcular c′ y el módulo volumétrico,
(c11 − c12). De modo que al calcular c′ y el módulo volumétrico,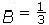 (c11 + 2c12), se obtienen los valores de c11 y c12.
(c11 + 2c12), se obtienen los valores de c11 y c12.
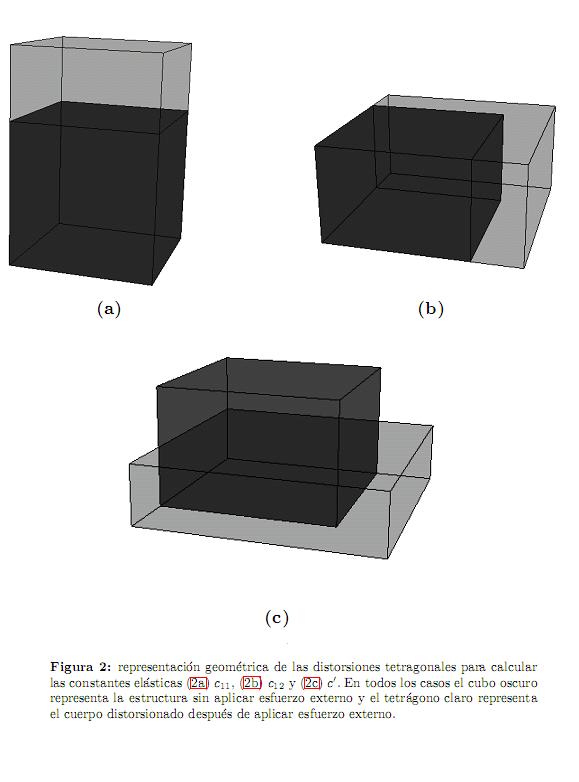
Similarmente, para calcular c44 se considera la distorsión ortorrómbica que también preserva el volumen (figura 3)

y la energía por celda unitaria asociada es

Es importante notar que hemos utilizado distorsiones que preservan el volu-
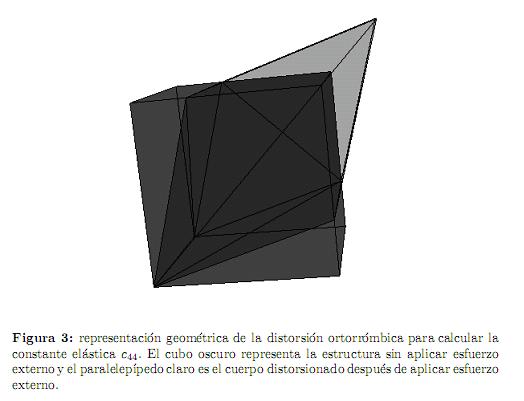
men para asegurar que el cambio en la energía total (ecuación (3)) es debido a las contribuciones elásticas y no por el cambio de volumen de la celda unitaria.
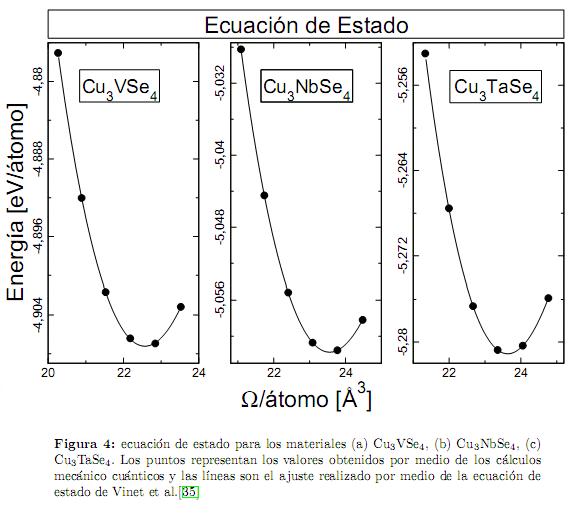
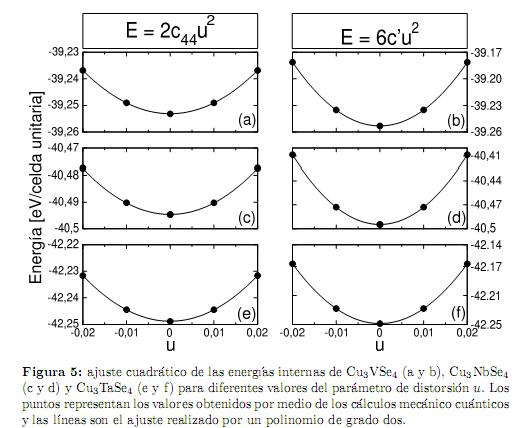
Inicialmente, nosotros calculamos la energía interna de cada compuesto como función del volúmen Ω de la celda unitaria (como se muestra en la figura 4, obteniendo de esta forma la curva E(Ω). Esta curva es ajustada por medio de la ecuación de estado universal para los sólidos propuesta por Vinet et al.[35], lo que nos permite encontrar el módulo volumétrico (B), su primera derivada (B′) y el volumen de equilibrio (Ω0). Seguidamente, para estos volúmenes de equilibrio se procedió a realizar las distorsiones descritas por (4) y (6), variando el parámetro de deformaciónucon valores 0, 0,01 y 0,02. La curvas obtenidas para E(u) son ajustadas con una función cuadrática como se muestra en la figura 5 para las distorsiones tetragonales y ortorrómbicas, respectivamente. Los coeficientes del término de segundo orden de las funciones cuadráticas obtenidas corresponden a las energías de las ecuaciones (5) y (7), con lo cual se obtienen las constantes elásticas c′ y c44. Finalmente, con el valor de B y c′ se obtienen c11 y c12.
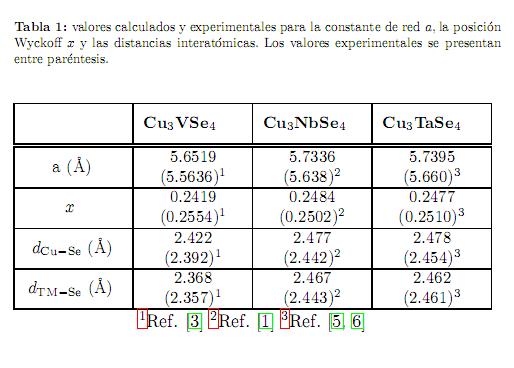
En la tabla 1 se muestran los valores calculados para la constante de red, el parámetro de Wyckoff aniónica y las distancias interatómicas de cada uno de estos compuestos. Se observa que los valores calculados de la constante
de red sobrestiman los valores experimentales en 1,6 %, 1,7 % y 1,4 % para Cu3VSe4, Cu3NbSe4y Cu3TaSe4, respectivamente, como es típico en cálculos con GGA. Para las distancias interatómicas Cu-Se (dCu−Se) calculadas, se observa un incremento a medida que se desciende por los elementos del metal de transición en la columna V-B de la tabla periódica, sobrestimando los valores experimentales en 1,2 %, 1,4 % y 1 %, para Cu3VSe4, Cu3NbSe4y Cu3TaSe4, respectivamente. Respecto a la distancia interatómica TM-Se (dTM−Se), se encuentra que los valores calculados sobrestiman los experimentales por 0,5 %, 1 % y 0,04 % para los materiales Cu3VSe4, Cu3NbSe4 y Cu3TaSe4, respectivamente.
En la tabla 2 se muestran los valores calculados para los monocristales de las sulvanitas del módulo volumétrico B, las constantes elásticas c11, c12 y c44 y el factor de anisotropía de Zener A. En la literatura no hay valores experimentales reportados para estas cantidades hasta el momento, así que
todos nuestros resultados son predicciones teóricas. Podemos concluir que el criterio de estabilidad de Born (c11 − c12 > 0, c11 + 2c12 > 0, c44 > 0 y c12 < B < c11)[36] se cumple para los tres compuestos, por lo tanto la estructura cristalina de las sulvanitas es mecanicámente estable bajo deformaciones elásticas. Se puede apreciar que el valor calculado de B, el cual mide la resistencia de un material a ser comprimido hidrostáticamente en respuesta a un cambio en la presión, disminuye cuando se desciende por los elementos de la columna V-B de la tabla periódica, y si comparamos estos valores de B con respecto al diamante (B= 442 GPa)[37], el cual es el compuesto más duro conocido, se concluye que estos materiales son muy compresibles. También se puede concluir que la constante elástica más rígida en los tres compuestos es c11, mientras que la más blanda es c12. La constante c44 también muestra ser
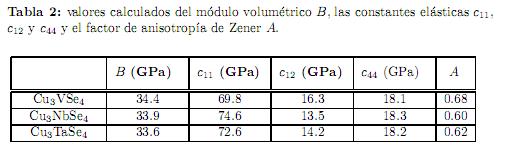
blanda, con valores aproximadamente iguales para los tres compuestos, muy cercanos a la vez de los valores de la constantec12 en todos los casos. Por lo tanto, estos compuestos son más rígidos ante deformaciones axiales, que basales o de cizalladura. Adicionalmente, el factor de anisotropía de Zener 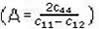 , establece que cuando A= 1 tenemos un cristal elásticamente
, establece que cuando A= 1 tenemos un cristal elásticamente
una descripción completa de las propiedades elásticas de estos compuestos. Estos pueden ser evaluados a partir de los valores de B, c11, c12 y c44 del monocristal usando el promedio de Voigt-Reuss-Hill[39, 40, 41]. Para calcular este promedio primero consideramos que un sólido policristalino esta formado por un gran número de granos (cristalitas) que pueden estar orientadas aleatoriamente (el policristal posee simetría esférica, grupo puntual ∞∞m) o todos los granos tienen la misma orientación (policristal con simetría puntual ∞∞). En cualquiera de los dos casos se puede considerar el material como un sólido isotrópico donde hay dos constantes elásticas independientes c11y c12. Sin embargo, es más común describir las propiedades elásticas de un sólido isotrópico en términos del módulo de cizalladura de Voigt GV , el módulo de cizalladura de Reuss GR, el módulo de cizalladura isotrópico G, el módulo de Young Y, la razón de Poisson ν y los coeficientes de Lamé µ y λ. Aplicando el método de Voigt-Reuss-Hill obtenemos las siguientes expresiones para los módulos de cizalladura, los cuales miden la respuesta del material ante una deformación ocasionada por una fuerza que actúa sobre el cuerpo, paralela a una de las caras, mientras que la cara opuesta permanece fija
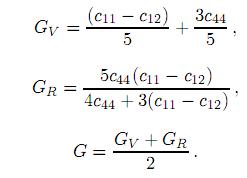
Aunque, el promedio de Reuss (límite inferior) y el promedio de Voigt (límite superior) no son enteramente correctos, el promedio de estos promedios es muy cercano a la situación real.
El módulo de Young caracteriza el comportamiento de un material elástico, según la dirección en la que se aplica una fuerza. Mide la facilidad o dificultad para estirar un material determinado, de modo que, un material con un módulo de Young bajo es un material fácil de estirar. Esta cantidad esta dada por
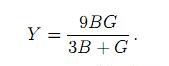
Si se somete el sólido a una tensión axial, éste se contrae lateralmente; mientras que si se le comprime, se ensancha lateralmente. La razón de Poisson, que es el cociente entre la deformación lateral y la deformación axial, es
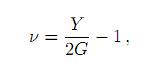
y cuantifica lo dúctil o quebradizo que es un material.
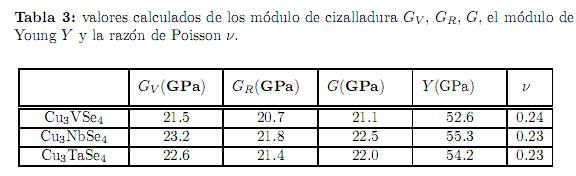
Los valores calculados para los módulo de cizalladura GV, GR, G, el módulo de Young Y y la razón de Poisson ν se muestran en la tabla 3. Tampoco existen valores experimentales reportados en la literatura para estas cantidades. Si comparamos estos valores calculados con un material bien estudiado tal como el vidrio (G= 24,1 GPa, Y= 60 GPa, ν = 0,23)[38] y considerando, como es aceptado empíricamente, que materiales con ν < 0,31 son quebradizos y materiales con ν > 0.31 son dúctiles; entonces podemos concluir que las sulvanitas evidencian un comportamiento frágil y quebradizo. Otro criterio utilizado para determinar el carácter dúctil o quebradizo de un material es la fórmula empírica de Pugh, la cual establece que si la razón entre B/G > 1,75 entonces el material se comporta de manera dúctil y en caso contrario el material es quebradizo. Los valores de esta razón para las sulvanitas están alrededor de 1,5 confirmando la naturaleza quebradiza indicada por la razón de Poisson.
Además, a partir de estas propiedades elásticas también se pueden encontrar otras cantidades, tales como la velocidad del sonido promedio para un material policristalino, dada por la siguiente expresión
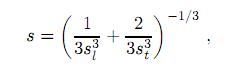
donde sl y st son la velocidades elásticas longitudinal y transversal del material policristalino, respectivamente. La onda transversal t es frecuentemente llamada onda de cizalladura, mientras la onda longitudinal l es llamada onda de compresión. Las velocidades de las ondas de cizalladura y de compresión se obtienen mediante las expresiones
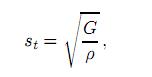
y
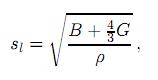
donde ρ es las densidad del cristal.
Finalmente, también hemos calculado la temperatura de Debye ΘD, que es básicamente una medida de la respuesta vibracional del material y está intimamente relacionada a varias propiedades termofísicas tales como la expansión térmica, la entropía vibracional, el calor específico y la temperatura de fusión. Además, la temperatura de Debye define la frontera entre el comportamiento clásico y cuántico de las vibraciones en un cristal. Por lo tanto, a bajas temperaturas las excitaciones vibracionales se derivan únicamente de vibraciones acústicas, este hecho implica que la temperatura de Debye calculada a partir de las constantes elásticas es la misma que la determinada de la medida del calor específico. Nosotros hemos calculado ΘD a partir de las constantes elásticas (usando la velocidad del sonido promedio) considerando la siguiente ecuación
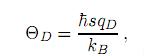
donde qD = 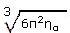 y ηα es la concentración atómica.
y ηα es la concentración atómica.
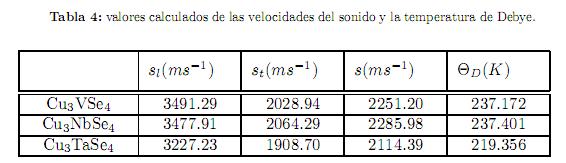
En la tabla 4 se muestran los valores encontrados para st, sl, s y ΘD. Los valores calculados de la temperatura de Debye nos indican un valor entre moderado y bajo de la conductividad térmica de estos compuestos. Desafortunadamente, hasta donde los autores conocen, no existen datos experimentales de éstas propiedades en la literatura para estos compuestos. Futuro trabajo experimental validará nuestros cálculos.
4 Conclusiones
En conclusión, hemos calculado las propiedades elásticas de los minerales de la familia isoestructural de las sulvanitas Cu3TMSe4 (TM = V, Nb, Ta), encontrando que son materiales compresibles pero que a la vez son frágiles y quebradizos. Más aún, en estos materiales es más difícil hacer deformaciones axiales que basales o de cizalladura, en cualquiera de las caras de la estructura cúbica. Por último los valores calculados de las constantes elásticas y las velocidades del sonido promedio transversal y longitudinal nos muestran un alto grado de anisotropía en estos compuestos.
Agradecimientos
Este trabajo fue financiado por el CODI-Universidad de Antioquia bajo el contracto No. IN558CE y el fondo de apoyo a trabajos de grado de la Universidad de Antioquia.
Referencias
1. Y.J. Lu and J.A. Ibers. Synthesis and Characterization of Cu3NbSe4 and KCu2TaSe4. Journal of Solid State Chemistry, ISSN 0022-4596, 107(1), 58-62 (1993). Referenciado en 133, 135, 142 [ Links ]
2. C. Mujica, G. Carvajal, J. Llanos and O. Wittke. Redetermination of the crystal structure of copper(I) tetrathiovanadate (sulvanite) Cu3VS4. Zeitschrift fur Kristallographie New Crystal Structures, ISSN 1433-7266, 213(1), 12 (1998). Referenciado en 133 [ Links ]
3. K. Klepp and D.Gurtner. Crystal structure of tricopper tetraselenidovanadate(V) Cu3VSe4. Zeitschrift fur Kristallographie New Crystal Structures, ISSN 14337266, 215(1), 4 (2000). Referenciado en 133, 135, 142 [ Links ]
4. M. Kars, A. Rebbah and H. Rebbah. Cu3NbS4Acta Crystallographica Section E ISSN 1600-5368, 61(8), i180 (2005). Referenciado en 133 [ Links ]
5. G. Delgado, A. Mora, S. Duran, M. Muñoz and P. Grima-Gallardo. Structural characterization of the ternary compound Cu3Ta Se4. Journal of Alloys and Compounds, ISSN 0925-8388, 439(1-2), 346-349 (2007). Referenciado en 133, 135, 142 [ Links ]
6. G. Delgado, A. Mora, P. Grima-Gallardo, S. Duran, M. Muñoz and M. Quintero. Synthesis and characterization of the ternary chalcogenide compound Cu3NbTe4. Chalcogenide Letters, ISSN 1584-8663, 6(8), 335-338 (2009). Referenciado en 133, 142 [ Links ]
7. D. Petritis, G. Martínez, C. Levy-Clement and O. Gorochov. Investigation of the vibronic properties of Cu3VS4, Cu3NbS4 and Cu3TaS4 compounds. Physical Review B, ISSN 1098-0121, 23(12), 6773-6786 (1981). Referenciado en 133 [ Links ]
8. Hervé Arribard and Bernard Sapoval. Theory of mixed conduction due to cationic interstitials in the p-type semiconductor Cu3VS4. Electrochimica Acta, ISSN 00 1-4686, 24(7), 751-754 (1979).Referenciado en 133 [ Links ]
9. D.M. Schleich and M. Rosso. Li+ insertion studies in Cu3VS4. Solid State Ionics ISSN 0167-2738, 5(1), 383 (1981). Referenciado en 133 [ Links ]
10. A. Golub, N. Allali, D. Guyomard and M. Danot. Lithium intercalation -deintercalation reactions using matrixes with the sulvanite structure: Dimensionality lowering of the host-structure Materials Research Bulletin, ISSN 0025-5408, 30(8), 959-966 (1995). Referenciado en 133 [ Links ]
11. G. Cressy, C. Henderson and G. van der Laan.Use of L-edge X-ray absortion spectroscopy to characterize multiple valence states of 3d transition metals; a new probe for mineralogical and geochemical research , Physics and Chemistry of Minerals, ISSN 0342-1791, 20(2), 111-119 (1993). Referenciado en 133 [ Links ]
12. D.A. McKeown, I.S. Muller, K.S. Matlack and I.L. Pegg. X-ray absorption studies of vanadium valence and local environment in borosilicate waste glasses waste glasses using vanadium sulfide, silicate, and oxide standards, Journal of Non-Crystalline Solids, ISSN 0022-3093, 298(2-3), 160-175 (2002). Referenciado en 133 [ Links ]
13. G. van der Laan, R. Pattrick, C. Henderson and D. Vaughan. Oxidation state variations in copper minerals studied with Cu 2p X-ray absorption spectroscopy Journal of Physics and Chemistry of Solids, ISSN 0022-3697, 53(9), 1185-1190 (1992). Referenciado en 133 [ Links ]
14. G. van der Laan and I.W. Kirkman. The 2p absorption spectra of 3d transition metal compounds in tetrahedral and octahedral symmetry, Journal of Physics: Condensed Matter, ISSN 0953-8984, 4(16), 4189-4204 (1992). Referenciado en 133 [ Links ]
15. J.W. Tate, P.F. Newhouse, R. Kykyneshi, P.A. Hersh, J. Kinney, D.H. McIntyre and D.A. Keszler. Chalcogen-based transparent conductors, Thin Solid Films, ISSN 0040-6090, 516(17), 5795-5799 (2008). Referenciado en 133, 134 [ Links ]
16. P.F. Newhouse, P.A. Hersh, A. Zakutayev, A. Richard, H.A.S. Platt, D.A. Keszler and J. Tate. Thin film preparation and characterization of wide band gap Cu3TaQ4(Q = S or Se) p-type semiconductors, Thin Solid Films, ISSN 00406090, 517(7), 2473-2476 (2009). Referenciado en 133, 134 [ Links ]
17. F. Zwick, H. Berger, M. Grioni, G. Margaritondo, L. Forró, J. LaVeigne, D. B. Tanner and M. Onellion. Coexisting one-dimensional and three-dimensional spectral signatures in TaTe4, Physical Review B, ISSN 1098-0121, 59(11), 77627766 (1999). Referenciado en 134 [ Links ]
18. N. Shannon and R. Joynt. The spectral, structural and transport properties of the pseudogap system (TaSe4)2I , Solid State Commun, ISSN 0038-1098 115(8), 411-415 (2000). Referenciado en 134 [ Links ]
19. S. Debus and B. Harbrecht. NbxTa7 S2(x=2.73), a structurally distinct (Nb, Ta)-rich sulfide obtaining its stability from the dissimilar cohesive energy of the two metals, Journal of Alloys and Compounds, ISSN 0925-8388, 338(1-2), 253-260 (2002). Referenciado en 134 [ Links ]
20. Y. Aiura, H. Bando, R. Kitagawa, S. Maruyama, Y. Nishihara, K. Horiba, M. Oshima, O. Shiino and M. Nakatake. Electronic structure of layered 1T-TaSe2 in commensurate charge-density-wave phase studied by angle-resolved photoemission spectroscopy, Physical Review B, ISSN 1098-0121, 68(7), 073408 073408-4 (2003). Referenciado en 134 [ Links ]
21. F. Dimroth and S. Kurtz. High-efficiency multijunction solar cells, Materials Research Society Bulletin, ISSN 0883-7694, 32(3), 230-235 (2007). Referenciado en 134 [ Links ]
22. Federico Capasso. Band-Gap Engineering: From Physics and Materials to New Semiconductor Devices, Science, ISSN 0036-8075, 235(4785), 172-176 (1987). Referenciado en 134 [ Links ]
23. W.F. Espinosa García, A.L. Morales Aramburo and J.M. Osorio Guillén. Electronic properties of the sulvanite compounds: Cu3TMS4(TM = V, Nb, Ta), Revista Colombiana de Física, ISSN 0120-2650, 40(1), 36-39 (2008). Referenciado en 134 [ Links ]
24. S. Adachi, Properties of Group-IV, III-V and II-VI Semiconductors. ISBN 0470-09032-4. John Wiley & Sons, West Sussex (2005) 134 [ Links ]
25. L. D. Landau and E. M. Lifshitz. Theory of elasticity. ISBN 075062633X. Butterworth-Heinemann, 3rd ed. New York (1986). Referenciado en 136 [ Links ]
26. L. Thomsen. Weak elastic anisotropy, Geophysics, ISSN 0016-8033, 51(10), 1954-1966 (1986). Referenciado en 136 [ Links ]
27. P. Hohenberg and W. Kohn. Inhomogeneous Electron Gas, Physical Review, ISSN 0031-899X, 136(3B), B864-B871 (1964). Referenciado en 137 [ Links ]
28. W. Kohn and L. J. Sham. Self-consistent equations including exchange and correlation effects, Physical Review, ISSN 0031-899X, 140(4A), A1133-A1138 (1965). Referenciado en 137 [ Links ]
29. J. Perdew, K. Burke and M. Ernzerhof. Generalized gradient approximation made simple, Physical Review Letters, ISSN 0031-9007, 77(18), 3865 3868 (1996). Referenciado en 137 [ Links ]
30. P. E. Blöchl. Projector augmented-wave method, Physical Review B, ISSN 01631829, 50(24), 17953-17979 (1994). Referenciado en 137 [ Links ]
31. G.B. Kresse and D.P. Joubert. From ultrasoft pseudopotentials to the projector augmented-wave method, Physical Review B, ISSN 1098-0121, 59(3), ISSN 17581775 (1999). Referenciado en 137 [ Links ]
32. G. Kresse and J. Furthmüller. Efficient iterative schemes for ab initio totalenergy calculations using a plane-wave basis set, Physical Review B, ISSN 10980121, 54(16), 11169-11186 (1996). Referenciado en 137 [ Links ]
33. H.J. Monkhorst and J.D. Pack. Special points for Brillouin-zone integrations, Physical Review B, ISSN 0163-1829, 13(12), 5188-5192 (1976). Referenciado en 137 [ Links ]
34. P. Söderlind, O. Eriksson, J. M. Wills and A. M. Boring. Theory of elastic constants of cubic transition metals and alloys, Physical Review B, ISSN 01631829, 48(9), 5844-5851 (1993). Referenciado en 137 [ Links ]
35. P. Vinet, J.R. Smith, J.H. Rose and J. Ferrante. Temperature effects on the universal equation of state of solids, Physical Review B.ISSN 0163-1829, 35(4), 1945-1953 (1987). Referenciado en 139, 140 [ Links ]
36. Cz. Jasiukiewicz and V. Karpus. Debye temperature of cubic crystals, Solid State Communications, ISSN 0038-1098, 128(5), 167-169 (2003). Referenciado en 141 [ Links ]
37. Marvin L. Cohen. Calculation of bulk moduli of diamond and zinc-blende solids Physical Review B. ISSN 0163-1829, 32(12), 7988-7991 (1985). Referenciado en 141 [ Links ]
38. James F. Shackelford, William Alexander and J. S. Park. CRC practical handbook of materials selection, ISBN 0849337097. CRC-Press, USA (1995). Referenciado en 144 [ Links ]
39. W. Voigt. Lehrbuch der Kristallphysik, ISBN 0384648401. Teubner, Leipzig, (1928). Referenciado en 143 [ Links ]
40. A. Reuss. Berechnung der Fliebgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle, Zeitschrift für Angewandte Mathematik und Mechanik, ISSN 0044-2267, 9 (1), 49-58 (1929). Referenciado en 143 [ Links ]
41. R. Hill. The Elastic Behaviour of a Crystalline Aggregate, Proceedings of the Physical Society A, ISSN 0370-1298, 65(5), 349-354 (1952). Referenciado en 143 [ Links ]

