










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkLuna Azul
versión impresa ISSN 1909-2474
Luna Azul n.30 Manizales ene./jun. 2010
ASSEMBLY AND VALIDATION BY THE METHOD OF COMBUSTION ANALYSIS AND NONDISPERSIVE INFRARED DETECTION FOR DETERMINING TOTAL ORGANIC CARBON (TOC) IN WATER
milton.rosero@ucaldas.edu.co
a Grupo de Cromatografía y Técnicas Afines GICTA, Universidad de Caldas.
b Grupo Estudio de Sistemas Contaminantes, Universidad de Nariño.
c Empresa de Obras Sanitarias de Pasto EMPOPASTO S.A. E.S.P.
Manizales, 2009-11-05 (Rev. 2010-05-18)
RESUMENLa técnica de análisis por combustión y detección por infrarrojo no dispersivo (IRND) fue validada para la determinación de materia orgánica en agua, cuantificada como carbono orgánico total (COT). Previamente se optimizó la eliminación del carbono inorgánico (CI) de la muestra con un tiempo óptimo de 1,5 min y una relación ácido de 5%. Se estableció un rango dinámico lineal (RDL) entre 3 y 20 mg/L de COT, en el cual la recta de regresión cumplió con los parámetros que acreditaron su linealidad según el análisis por el método de los mínimos cuadrados, mostrando un coeficiente de correlación de 0,9994. La sensibilidad expresada por la pendiente de la recta de regresión indicó una variación de aproximadamente 5 unidades en la respuesta del detector por cada mg/L de COT. Los límites de detección y cuantificación obtenidos a partir de la recta de regresión fueron 0,517 y 1,722 mg/L de COT, respectivamente. La precisión de la técnica, teniendo como meta un 5% en coeficiente de variación (CV), fue mejor en el agua potable y en concentraciones cercanas a ésta (aprox. 7 mg/L de COT), mientras que las mayores desviaciones se presentaron en concentraciones cercanas a los límites inferior y superior del RDL. Las recuperaciones de concentraciones conocidas sobre muestras reales adicionadas fueron del 85% con baja adición y del 83% con adición alta, valores que sugieren una reevaluación del desempeño de la técnica con respecto a su exactitud, no obstante se alcanzó la meta de recuperación comprendida entre el 70 y el 130%. La técnica así establecida es por tanto apta para el análisis de aguas crudas, potables y residuales y es viable su utilización en el seguimiento a procesos de oxidación avanzada aplicados en el tratamiento de agua para consumo humano.
PALABRAS CLAVE:Validación, carbono orgánico total, homocedasticidad, límite de detección, límite de cuantificación.
ABSTRACTThe technique of combustion analysis and detection by non-dispersive infrared (NDIR) was validated for the determination of organic matter in water, quantified as total organic carbon (TOC). Previously optimized the elimination of inorganic carbon (IC) of the sample with an optimum time sparge of 1.5 min and an acid ratio of 5%. It established a linear dynamic range (LDR) between 3 and 20 mg/L of TOC, in which the regression line that met the parameters credited its linearity as analyzed by the method of least squares, showing a correlation coefficient of 0,9994. The sensitivity expressed by the slope of the regression line indicated a variation of about 5 units in the detector response for each mg/L of TOC. The detection and quantification limits obtained from the regression line were 0.517 and 1.722 mg/L of TOC, respectively. The precision of the technique, aiming at 5% coefficient of variation (CV) was better in the drinking water at concentrations close to it (about 7 mg/L of TOC), whereas larger deviations were presented at concentrations near the lower and upper limits of LDR. The recoveries of known concentrations of actual samples of 85% additions were low addition and 83% with high added values that suggest a reevaluation of the performance of the technique with respect to its accuracy, however the goal was reached between recovery 70 to 130%. The technique is well established thereby capable of analyzing raw water, drinking and waste and feasible to use in monitoring advanced oxidation processes applied in the treatment of drinking water.
KEY WORDS:Validation, Total Organic Carbon, Homoscedasticity, Detection Limit, Quantification Limit.
INTRODUCCIÓN
La materia orgánica natural (MON) está presente en todas las aguas superficiales y en las aguas relativamente poco profundas. Se origina en el suelo donde se han acumulado materias vegetales finamente divididas y en descomposición. Parte viene proporcionada también por la digestión de los animales. Los fondos sumergidos o periódicamente recubiertos son lugares de una vida activa que proporciona una gran variedad de productos orgánicos complejos. Además están los productos del metabolismo de las algas y de los hongos que son los responsables de los sabores y olores en el agua[1].
Las aguas con importante concentración de materia orgánica de procedencia natural, son a menudo ácidas y están muy poco mineralizadas, ya que provienen de terrenos cuarzosos o mineralizados. Las aguas de regiones de turba están muy cargadas de materia orgánica y tienen un notable color marrón rojizo[2].La MON está formada generalmente, por sustancias químicas que son polielectrolitos aniónicos de bajo a moderado peso molecular, cuya carga se debe principalmente a los grupos carboxílico y fenólico[3, 4, 5]. Dichas sustancias pueden ser ácidos fúlvicos (más solubles) y ácidos húmicos (menos solubles).
En términos generales, esta materia orgánica se expresa como contenido o concentración de carbono orgánico, y haciendo la aclaración de que "ninguno de los métodos de demanda de oxígeno da una estimación precisa del carbono orgánico disuelto en el agua"[1], se hace imprescindible utilizar una técnica específica para determinar este parámetro.La técnica utilizada es la combustión total de la materia orgánica a CO2 y detección de este producto con detector infrarrojo no dispersivo. La muestra se prepara por acidificación y aireación para remover el carbono inorgánico, luego se inyecta la muestra tratada en un horno donde se evapora el agua y el carbono orgánico hace combustión catalítica a CO2, el cual es transportado por una corriente de aire UP hasta el detector, cuya señal es una variación de voltaje proporcional a la concentración de carbono orgánico total (COT).
El interés en el análisis y cuantificación del COT, radica en que este parámetro se utiliza comúnmente como indicador de la calidad del agua cruda destinada al tratamiento de potabilización y como indicador de la materia orgánica que permanece remanente después de los procesos destinados a su eliminación durante el proceso de potabilización y se constituye como precursora en la formación de subproductos de desinfección (SPDs) al entrar en contacto con el cloro aplicado como desinfectante[5-14].El aporte de carbono total (CT) en muestras de agua lo hacen tanto sustancias orgánicas como inorgánicas, por tanto se debe distinguir el carbono orgánico total (COT) del carbono inorgánico (CI). El CI consiste netamente en carbonatos y en CO2 disuelto en agua. A su vez, el COT está compuesto por dos porciones: el carbono orgánico volátil o purgable (COP) y el carbono orgánico no volátil o no purgable (CONP). Si la porción orgánica volátil es despreciable, se asume que el CONP equivale al COT. El COT se puede medir usando uno de los 3 métodos siguientes[9, 15, 16]:
CT-CI: se determina por separado en la muestra el CT, luego el CI y por diferencia entre ambos parámetros se calcula el COT.CONP: la muestra se acidifica con HCl hasta un pH igual o menor a 3 de manera que todos los carbonatos y bicarbonatos se transformen en CO2, el cual junto con el CO2 disuelto se volatiliza al burbujear la muestra con aire libre de CO2, de esta manera se elimina el componente inorgánico, mientras que el CT restante se mide y corresponde al CONP que es igual al COT. Este método es el más utilizado para determinar COT y se aplica si la porción orgánica volátil es despreciable, como en el caso de las aguas destinadas al consumo humano.
COP+CONP: se utiliza cuando la cantidad de COP no es despreciable en la muestra.En este estudio se optimizó y validó la técnica de análisis por combustión y detección por IRND para determinación de COT utilizando el método de medición directa del CONP, al considerar que este método introduce menos errores que la determinación por separado de CT y CI, además se supone que en la matriz de agua cruda destinada para potabilización (matriz de interés), se puede despreciar la porción orgánica volátil.
MATERIALES Y MÉTODOSEl equipo utilizado fue un analizador de carbono orgánico total Shimadzu TOC-V CSN equipado con detector infrarrojo no dispersivo IRND. El inyector automático del equipo está dotado con una válvula de 8 puertos. La reacción de combustión se realiza en un tubo de cuarzo a 680 °C y es catalizada con un catalizador regular de platino (recomendado para concentraciones mayores a 0,5 mg/L de C). La presión de trabajo fue de 43 psi y se utilizó aire UP como gas de arrastre con un flujo de 150 ml/min.
Materiales • Balones aforados clase A de 50, 100, 250 y 1000 ml
• Pipetas aforadas clase A de 2, 5, 10, 25, y 50 ml
• Beakers de 250 ml
• Viales ámbar de 40 ml con tapa PTFE
• Frasco lavador
El lavado del material de vidrio se realizó con una solución diluida de TEXAPON, se enjuagó con abundante agua de la llave y posteriormente con agua destilada tipo II. Finalmente, todo el material se purgó con agua para reactivos tipo ASTM I y se dejó secar a temperatura ambiente.
Reactivos • Ftalato ácido de potasio KC8H5O4
• Agua para reactivos (agua ASTM I)
• HCl 2 M
• H3PO4 25%
• Aire libre de CO2 (UP)
A partir de la solución de CT se preparó una solución de concentración 20 mg/L de C con la cual se elaboró la curva de calibración en un rango de 5 a 20 mg/L utilizando la función de autodilución del equipo. Esta curva se utilizó para cuantificar el CONP en los diferentes ensayos de optimización del tiempo de purga y relación de HCl. De la misma manera se elaboró la curva de calibración con la que se validó el método.
Para la validación se preparó una solución intermedia de 100 mg/L de C que se almacenó en un frasco de vidrio ámbar y se utilizó para preparar los estándares y dopar las muestras utilizadas en esta etapa.El COT se determinó por medida directa del equipo como carbono orgánico no purgable (CONP). La muestra, de la cual se requiere como mínimo 15 ml de muestra, debe agitarse antes del análisis. La manguera de introducción de muestra al equipo se ubica en el seno del líquido.
La validación se realizó analizando por duplicado durante seis días consecutivos las muestras cuyas denominaciones y características se presentan en la Tabla 1. El blanco fue agua destilada tipo I con posterior tratamiento mediante ósmosis inversa. Las muestras reales M1 y M2 a las que hace referencia la Tabla 6 fueron muestras compuestas pertenecientes a las Empresas Públicas de Armenia (EPA), que se tomaron en un lapso de 6 horas, cuyo volumen final de 4 L se guardó refrigerado durante los días de la validación. Las muestras adicionadas, lo mismo que los estándares, se prepararon diariamente para los diferentes ensayos.

Los resultados de esta etapa se procesaron mediante un aplicativo de Excel del GEMS del Water Research Center del Reino Unido, obteniéndose la estimación de los atributos de validación: precisión, exactitud y recuperación.
RESULTADOS Y DISCUSIÓN Linealidad
De acuerdo a las especificaciones del fabricante del equipo, en las curvas de calibración para CT se observó comportamiento lineal de la técnica desde 1 hasta 1000 mg/L de C. Éste es un rango bastante amplio que permite procesar muestras con bajo contenido de carga orgánica, como por ejemplo agua potable proveniente de plantas que cuentan con tratamientos avanzados de potabilización, así como muestras de aguas residuales tanto domésticas como industriales. Sin embargo, para aumentar la sensibilidad del análisis y de acuerdo a la naturaleza de las muestras a procesar se requiere que se realicen diferentes curvas que comprendan el rango en el que se desea trabajar.
Al graficar los valores de área bajo la curva con respecto a la concentración de cada estándar se obtuvo la recta que se presenta en la Figura 1, la cual muestra la relación de tipo lineal en el intervalo de concentraciones en estudio. El coeficiente de correlación R² = 0,9966 es aceptable, pero teniendo en cuenta que la curva es elaborada por el equipo se esperaría un valor de 0,9990 o mayor, por lo cual se propuso la optimización de los factores que inciden grandemente en la eliminación del carbono inorgánico, a saber: relación de ácido y tiempo de burbujeo.
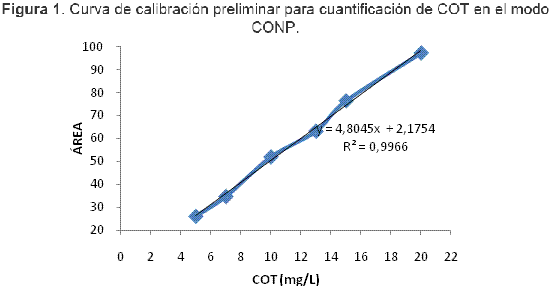
Optimización del proceso de eliminación de CI para cuantificación de COT en el modo CONP
Las diferentes combinaciones de los niveles escogidos, obtenidas a partir de la matriz que se indicó en la Tabla 7 y los resultados obtenidos por duplicado tanto para concentración como para recuperación se muestran en la Tabla 2.
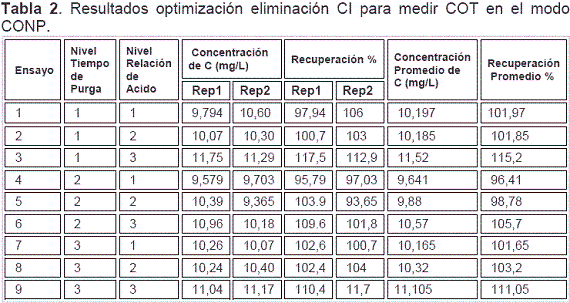
La Tabla 3 presenta el análisis de varianza, ANOVA, el cual descompone la variabilidad de la recuperación debida a las contribuciones de ambos factores. La contribución de cada factor se determinó eliminando los efectos causados por el otro factor.
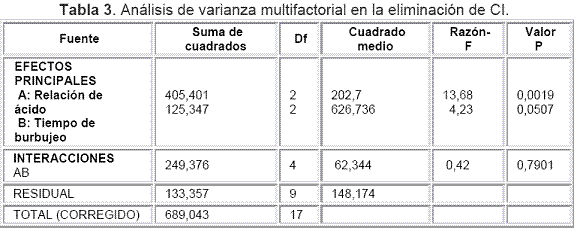
Según los resultados de la Tabla 3, se concluyó que no hay interacción interfactorial entre los factores Relación de ácido y Tiempo de burbujeo, pues el valor P para la interacción de los dos factores es mayor a 0,05. Sin embargo, independientemente los factores Relación de ácido y Tiempo de burbujeo tienen peso estadístico pues su valor P es menor de 0,05 para el primer factor y es igual a 0,05 para el segundo. Indiscutiblemente, la relación de ácido tendría mayor peso, ya que si se logra acidificar el medio eficientemente, se llevará todos los bicarbonatos y carbonatos a CO2, eliminando al máximo el componente inorgánico de las muestras. Por tanto, estos factores se analizaron separadamente, aplicando un proceso de comparación múltiple para determinar cuáles medidas eran estadísticamente diferentes unas de otras, de manera que se evaluaron los promedios de recuperación y los intervalos al 95% obtenidos para el Tiempo de burbujeo y Relación de ácido.
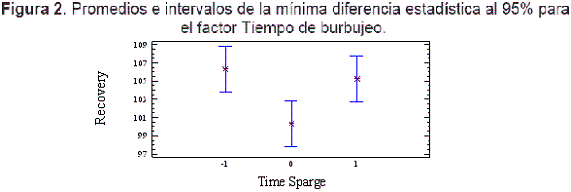
Para el factor Tiempo de burbujeo, los niveles -1 y 1 generaron recuperaciones estadísticamente equivalentes, como se puede observar en la Figura 2 y se eligió al nivel -1 (1,5 min), por ser el menor tiempo, como nivel óptimo para éste factor.
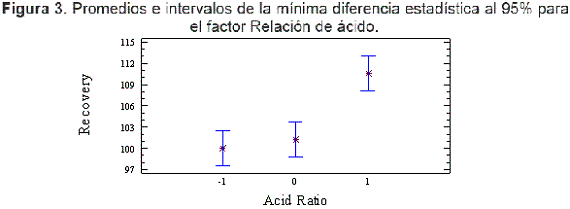
La Figura 3 muestra un diagrama de promedios e intervalos comparando los resultados obtenidos a tres niveles del factor Relación ácido. Los resultados demostraron que la diferencia estadística entre los niveles -1 y 0 era baja, si bien las condiciones de 1 difieren de las de -1 y 0. Al haber obtenido una mejor recuperación en el nivel 1, se eligió este nivel 1 (5,0 %) como el óptimo para el análisis.
Rango dinámico lineal
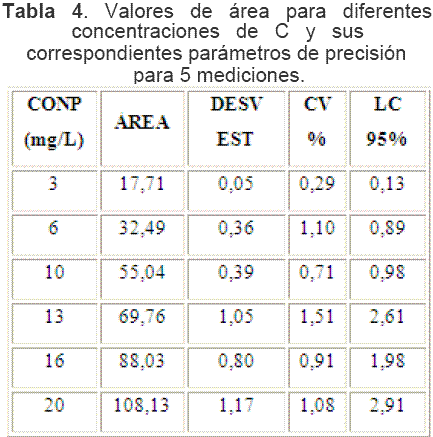
La Tabla 4 muestra los resultados de una curva de calibración (en adelante denominada curva de trabajo) que se realizó empleando los niveles óptimos de los factores Relación de ácido y Tiempo de burbujeo.
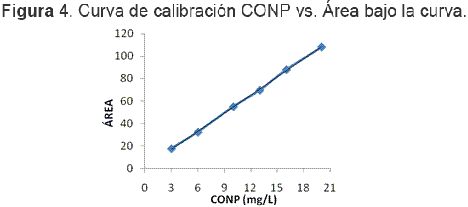
La prueba de hipótesis de linealidad comprobó que hay correlación significativa entre las variables x e y. Según el ajuste por el método de los mínimos cuadrados de la anterior curva de calibración, se obtuvo un coeficiente de correlación r2 de 0,9994, el mejoramiento de la linealidad es consecuencia neta de la optimización del proceso de medición de COT por el modo CONP y la recta queda descrita mediante la ecuación y = 5,3676(+0,0654)x + 1,0267(+0,8321). En la Figura 4 se puede observar esta curva.
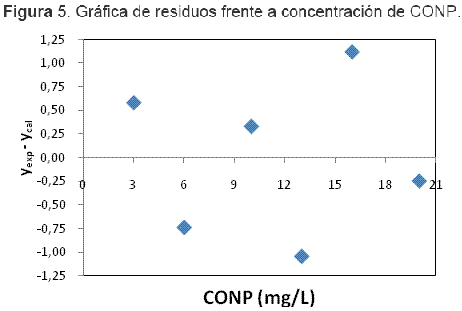
El comportamiento de los residuos de respuesta frente a sus valores estimados fue aleatorio con respecto a las concentraciones, como se puede observar en la Figura 5, cumpliendo con el criterio de homocedasticidad requerido en este tipo de regresiones. Además, la máxima desviación en estos residuos fue de 1.
Sensibilidad
La sensibilidad de la técnica, determinada por la pendiente de la curva de calibración: 5,3676(+0,0654), indica que una variación de 1 mg/L de CONP origina un cambio aproximado de 5,3 unidades en la respuesta del detector, sensibilidad suficiente para el análisis de muestras de agua potable.
Los valores calculados para estos límites a partir de la recta de regresión, se presentan en la Tabla 5. El límite de detección cumple con el requerimiento de ser como máximo, igual al 10% del valor guía para agua potable que para Colombia se ha establecido en 5 mg/L de C.

Se puede asumir que los límites instrumentales de detección y de cuantificación, son muy cercanos a los límites de detección y de cuantificación del método, respectivamente, sabiendo que la matriz a trabajar es sencilla y libre de interferencias[8].
ETAPA DE VALIDACIÓN Precisión
El análisis de varianza de un factor ANOVA para cada una de las soluciones trabajadas en la etapa de validación se presenta en la Tabla 6. La hipótesis nula Ho indica que la varianza entre lotes y dentro de lotes es igual y la hipótesis alterna H1 plantea que las varianzas entre lotes y dentro de lotes son diferentes. Para el contraste de estas hipótesis se empleó el estadístico F teniendo en cuenta que si F calculado es mayor al F teórico se rechaza Ho y se acepta la H1.
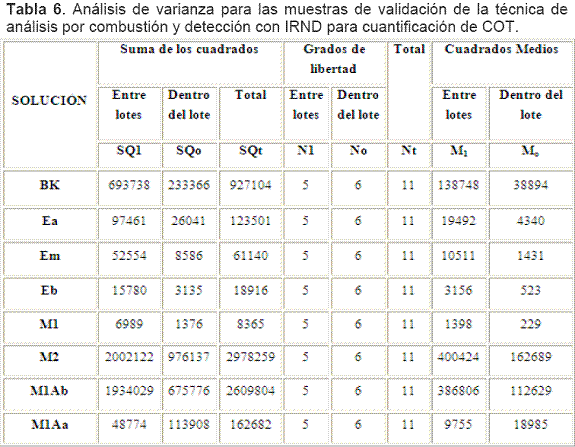
La Tabla 7 presenta el resumen de las desviaciones estándar, los promedios de las concentraciones y sus coeficientes de variación, como parámetros indicativos de precisión.
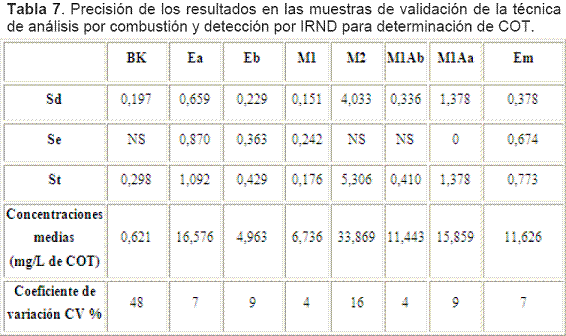
Para las soluciones: Blanco, M2 y M1Ab, la variabilidad es no significativa (NS) y muy probablemente obedezca a errores de tipo aleatorio pues se retuvo la Ho al 95% y no se pudo demostrar que era falsa, así las varianzas entre días y entre replicas son iguales o mejor aún su diferencia es no significativa.
Para las soluciones: Ea, Em, Eb y M1 las variabilidades son significantes, lo que probablemente se deba a errores de tipo aleatorio y sistemático pues se rechazó la Ho y se acepto la Ha al 95%, de manera que las varianzas entre días y entre replicas resultan diferentes, es decir que su diferencia es estadísticamente significativa.Según el criterio del CV cumplen la meta (<5%) en primera instancia las soluciones M1 y M1Ab. Según el método del WRC cumplen la meta de variabilidad en segunda instancia las soluciones Ea y Em.
Definitivamente las soluciones Eb, M2 y M1Aa no cumplieron la meta incluso ni en segunda instancia, el valor de CV de 9, 16 y 9%, respectivamente, advertía este comportamiento, luego estas soluciones deberían repetirse para asegurar su cumplimiento.
RECUPERACIÓN Y EXACTITUDEn general, las recuperaciones decrecieron conforme transcurrieron los días de la validación, por lo que se puede suponer una baja eficiencia en la oxidación del carbono a CO2, hecho que directamente depende del estado del catalizador, así como del Tiempo de burbujeo y la Relación ácido pese a que estos factores se hayan optimizado, por lo que deben ser evaluados continuamente.
La Tabla 8 resume las recuperaciones obtenidas en las muestras adicionadas, con lo que se hizo una estimación de la exactitud del método.
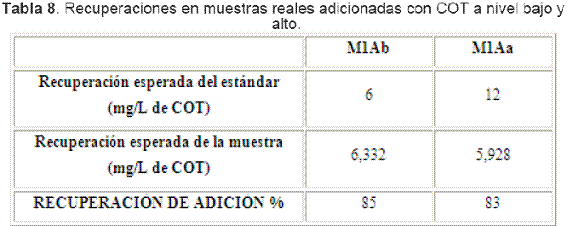
En ninguna de las adiciones se cumplió la meta de recuperación que debe ser mayor a 90%, lo que sugiere inicialmente que la metodología muestra una exactitud media no aceptable para análisis de CONP por análisis de combustión con detector IRND en aguas de consumo. Por otra parte, de acuerdo a la US EPA[19], que propone valores de recuperación entre 70 y 130%, el método en estudio sí sería aceptable para el análisis de CONP. Sin embargo, se puede hacer una revisión de los rangos de recuperación aceptables para este tipo de análisis, ya que no hay acuerdo entre investigadores ni entes afines a metrología.
Además, el hecho de que se hayan obtenido bajas recuperaciones también sugiere la posible presencia de sustancias interferentes en la matriz (agua tratada), como por ejemplo cloruros; en tal caso, la oxidación de Cl- a Cl2 gaseoso disminuye la eficiencia de la reacción de oxidación de la materia orgánica presente en la muestra y por tanto disminuye la señal. Prueba de ello, sería el estado del scrubber (absorbedor de halógenos) que presentaba hasta ese momento un color verde, característico de la presencia de Cl2 gaseoso.
CONCLUSIONES• Se validó el método de combustión y detección por IRND para determinación de COT en agua, al haber cumplido la técnica con los atributos de validación requeridos para este análisis, como fueron linealidad, sensibilidad y bajos límites de detección y cuantificación; sin embargo, los parámetros de precisión y exactitud deben ser reevaluados para corroborar el cumplimiento de las metas propuestas sabiendo que se pueden adoptar metas más permisivas.
• El rango dinámico lineal entre las concentraciones 3 a 20 mg/L de COT resultó eficiente para análisis de agua potable con el método de combustión y detección por IRND; no obstante, para análisis de aguas crudas se debe ampliar este rango a fin de evitar el error sistemático que se estaría presentando cuando el equipo debe realizar dilución de la muestra.• Es posible que la baja precisión que se presentó en la muestra de agua cruda durante la validación, se deba a que como ésta es una muestra concentrada y por lo general la respuesta excedía el RDL, el equipo debía volver a tomar muestra y hacer purga, por lo que la última inyección generaba mayor desviación, de manera que es más recomendable diluir la muestra manualmente.
• El hecho de que las soluciones Eb y M1Aa no hayan cumplido con la meta de precisión sugiere que las concentraciones cercanas a los límites superior e inferior del RDL trabajado pueden presentar falta de precisión, por lo tanto, según el tipo de muestras a analizar se debe recalibrar de manera que se asegure una ubicación intermedia de las respuestas en el RDL.
AGRADECIMIENTOSLos autores agradecen la cofinanciación de la Universidad de Nariño y de la Empresa de Obras Sanitarias de Pasto EMPOPASTO S.A. E.S.P. De igual manera, agradecen la colaboración del Departamento de Química de la Universidad del Quindío en donde fue montada la técnica, y la colaboración especial de la Química Cristina León Salazar.
REFERENCIAS BIBLIOGRÁFICAS
1. Revee, Roger N. (2002). Introduction to environmental analysis. Inglaterra: John Wiley & Sons, Ltd. [ Links ]
2. Gomella, C., Guerree, H. (1977). Tratamiento de aguas para abastecimiento público. Barcelona: editores técnicos asociados, S.A. [ Links ]
3. Snoeyink, Vernon; Illinois, Urbana; Jenkins, David. (1994). Química del agua. Berkeley: LIMUSA. [ Links ]
4. Colin, Baird. (2001). Química Ambiental. Madrid: Ed. Reverté S.A. [ Links ]
5. Villegas, C. 2005. Evaluación de la formación potencial de trihalometanos y trihalometanos en agua potable, usando microextracción en fase sólida como método de extracción. Bogotá. Septiembre 2009 Disponible en: http://www.uninorte.edu.co/divisiones/Ingenierias/IDS/index.asp. [ Links ]
6. Wong, H., Mok, K.M., Fan, X.J. (2007). Natural organic matter and formation of trihalomethanes in two water treatment processes. Desalination, 210, 44-51. [ Links ]
7. Sirivedgin, T., Gray, K. (2005). Comparison of the disinfection by-product formation potentials between a wastewater effluent and surface waters. Water Research, 39, 1025-1036. [ Links ]
8. Rosero, M., Latorre, J., Torres, W., Delgado, L. (2004). Presencia de materia orgánica y subproductos de la desinfección con cloro. Caso sistema de tratamiento de agua para consumo humano, Puerto Mallarino, Cali - Colombia. Tesis de Maestría. Universidad del Valle, Cali. [ Links ]
9. Wallace, B., Purcell, M., Furlong, J. (2002). Total organic carbon analysis as a precursor to disinfection byproducts in potable water: Oxidation technique considerations. J. Environ. Monit., 4, 35-42. [ Links ]
10. Gallard, H., Gunten, U. (2002). Chlorination of natural organic matter: kinetics of chlorination and of THM formation. Water Research, 36, 65-74. [ Links ]
11. Gang, D., Clevenger, T.E., Banerji, S.K. (2003). Relationship of chlorine decay and THMs formation to NOM size. Journal of Hazardous Materials, A96, 1-12. [ Links ]
12. Liu, W., Cheung, L., Yang, X., Shang, C. (2006). THM, HAA and CNCl formation from UV irradiation and chloramination of selected organic waters. Water Research, 40, 2033-2043. [ Links ]
13. Sorlini, S., Collivignarelli, C. (2005). Trihalomethane formation during chemical oxidation with chlorine, chlorine dioxide and ozone of ten Italian natural waters. Desalination, 176, 103-111. [ Links ]
14. Uyak, V., Ozdemir, K., Toroz, Ismail. (2007). Multiple linear regression modeling of disinfection by-products formation in Istanbul drinking water reservoirs. Science of the Total Environment, 378, 269-280. [ Links ]
15. Shimadzu Corporation. (2001). Manual de instrucciones TOC-V CSN SHIMADZU. [ Links ]
16. American Public Health Association, American Water Works Association y Water Pollution Control Federation Standard Methods for the Examination of Water and Wastewater. (1994) 14th, edition. Mét. 5310B. [ Links ]
17. EPA Method 415.1 (Combustion or Oxidation). Total Organic Carbon in Water. [ Links ]
18. Instituto de Hidrología, Meteorología y estudios ambientales IDEAM. Gustavo Alfonso Coy. (1999). Protocolo estandarización de métodos analíticos. Bogotá. [ Links ]
19. EPA, QA/QC. (1995). Guidance for sampling and analysis of sediments, water, and tissues for dredged material analysis. [ Links ]

