











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkI. INTRODUCCIÓN
Para la ciencia biológica y médica, el alineamiento de las secuencias de moléculas como proteínas y ácidos nucleicos (ADN y ARN), es un tópico de investigación de mucho interés por parte de la comunidad científica, ya que la secuencia de estas moléculas establece la base de la vida misma. Las secuencias encontradas en el ADN son utilizadas por investigadores en biología molecular y genética, ciencia forense y médica. Llevando a cabo un proceso de comparación o alineamiento de dos o más secuencias, se pueden identificar desde los genes que corresponden a enfermedades graves y desórdenes genéticos, hasta llegar a identificar la presencia de un sospechoso en la escena de un crimen.
El proceso de alineamiento está basado en algoritmos de programación dinámica o heurísticos cuyo objetivo es el de encontrar regiones iguales y diferentes a lo largo de toda la cadena de moléculas. Según el tipo de comparación, los algoritmos son clasificados en locales y globales. Cada tipo genera distintas variables de costo-beneficio sobre el tiempo de computación y memoria usada; sin embargo el problema de interés principal se centra en el hecho que las secuencias tienen una gran cantidad de bases o aminoácidos (alrededor de tres mil millones para los genomas más grandes) y dichas bases son muy variables entre sectores, lo cual implica mayor tiempo de procesamiento y demasiados recursos de computación.
Con el propósito de mitigar el problema anterior, en la literatura se encuentran varios tipos de implementación en software y/o hardware de los algoritmos de alineamiento como los presentados en [1,2,3,4].
Este artículo presenta la implementación en hardware del primer proceso del algoritmo de alineamiento de Needleman-Wunsch basado en programación dinámica, es decir, el cálculo de la matriz de puntuación. En este caso, se implementa el algoritmo k-band, el cual es una modificación del algoritmo de alineamiento global propuesto por primera vez en 1970, por Saul Needleman y Christian Wunsch. La arquitectura hardware es basada en un arreglo paralelo y es descrita por medio de lenguaje VHDL. La arquitectura diseñada puede ser usada para alinear grandes secuencias disponiendo de un dispositivo FPGA con una gran cantidad de recursos o haciendo uso de arreglos matriciales de FPGAs. Este diseño hardware también puede ser usado para procesar otros algoritmos basados en programación dinámica, modificando la función de maximización en el elemento de procesamiento. El artículo está organizado de la siguiente manera: La sección 2 presenta los trabajos previos. Sección 3 describe el algoritmo de Needleman-Wunsch (NW) y la modificación k-band. Sección 4 describe la arquitectura hardware propuesta. La sección 5 presenta las simulaciones software y la verificación del diseño. Conclusiones y trabajo futuro son presentados en la sección 6.
II. TRABAJOS PREVIOS
En la literatura se encuentran diversos trabajos relacionados con el uso de la programación dinámica en algoritmos para estudiar la genética, desde su primer resultado a mediados de los 70's (veinte años después de la primera lectura de una cadena proteínica). Estos algoritmos fueron usados debido a que es muy difícil de realizar manualmente una comparación y mucho menos el alineamiento dos secuencias con millones y millones de bases. Cuatro décadas más tarde sigue siendo necesario realizar una mayor investigación sobre los algoritmos utilizados para dicho fin, como lo menciona L. Alimehr en su trabajo dedicado al desempeño de los tipos de algoritmos de alineación e incluso él presenta técnicas globales de alineamiento [1].
Por otra parte se presentan aceleradores basados en hardware para resolver el problema del alineamiento usando algoritmos heurísticos. El desarrollo de estos aceleradores aumenta con la culminación del proyecto del genoma humano en el 2003. En [2], M. Kim presenta el diseño de un sistema basado en un comparador y un alineador implementados en FPGA para ayudar al software a calcular las tablas de puntuación en el alineamiento de cadenas de nucleótidos, realizando el procedimiento de forma paralela sobre los miles de millones de bases. En [3] se presenta un trabajo de tesis que consiste en la implementación de aceleradores para el algoritmo Smith-Waterman en FPGA.
En [4], los autores presentan un acelerador en hardware basado en FPGAs para el alineamiento de cadenas de ADN. En dicho trabajo, el objetivo es mostrar el alto desempeño que tienen los algoritmos de programación dinámica en los dispositivos de hardware programable donde el diseño es programado con OpenCL. En [5] se presenta la implementación del algoritmo de Smith-Waterman para el alineamiento local usando una arquitectura paralela.
III. ALGORITMO DE NEEDLEMAN-WUNSCH
En el algoritmo de Needleman-Wunsch [6], usado para alinear dos secuencias de ADN, primero se debe crear una matriz bidimensional de celdas con tamaño igual al producto de la longitud de cada secuencia (MxN); segundo, calcular en cada celda el máximo valor de puntuación entre los valores de las tres celdas alrededor y generar el puntero de la comparación para conocer la celda que generó dicho valor máximo; tercero, encontrar el camino de regreso (trace-back process) desde la última celda hacia la primera celda para encontrar la solución óptima de alineamiento [8,9].
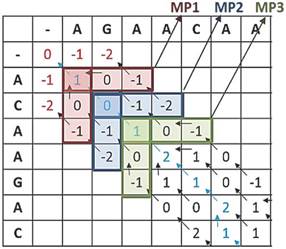
La Fig. 1 muestra una matriz ejemplo tomada de [7], la cual contiene sus valores de puntuación y los punteros utilizados en el proceso de trace-back.
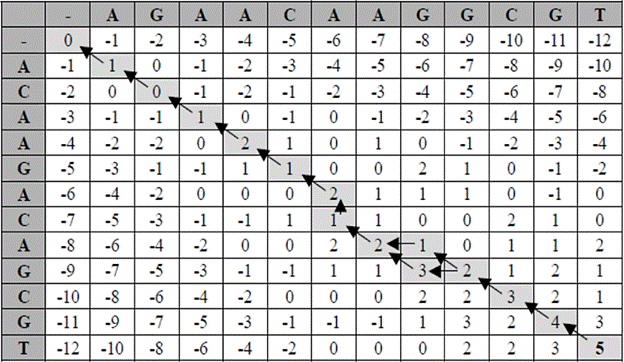
Desde la Fig. 1 podemos observar que el alineamiento óptimo tiene 2 caminos los cuales poseen sólo un salto por fuera de la diagonal principal. Cada camino genera los siguientes alineamientos, respectivamente:
X: ACAAGACA-GCGT
Y: AGAACA-AGGCGT
X: ACAAGACAG-CGT
Y: AGAACA-AGGCGT
Como se puede observar, en ambas alineaciones se presenta la misma cantidad de aciertos, inserciones y supresiones (GAPs), por lo que ambos caminos tendrán la puntuación máxima posible en el alineamiento.
En el algoritmo 1 se presenta el pseudocódigo para calcular el valor de puntuación para cada celda desde la coordenada (0,0) hasta (M,N):

En resumen, el algoritmo primero inicializa la primera fila y la primera columna considerando un conteo inverso que reduce el valor del gap (d) en las siguientes celdas. Para el resto de las filas se debe tener en cuenta el máximo entre el puntaje de la celda de arriba menos el gap, el puntaje de la celda izquierda menos el gap y el puntaje de la celda diagonal más una función s(xi,yi) que corresponde a los puntajes de acierto o diferencia entre los dos nucleótidos correspondientes a las posiciones i y j. Evaluando cada celda, la tabla se va llenando hasta quedar completa y se deben almacenar los valores de cada puntaje y los punteros de donde proviene su valor (izquierda, derecha, diagonal).
Este algoritmo requiere una gran cantidad de procesamiento y de memoria para el cálculo de los valores de puntuación y los punteros de cada celda. En este caso, para cada par de secuencias se deben realizar NxM cálculos, entonces en cadenas de genomas que tienen un orden de decenas de millones de pares, la matriz tendrá un número de celdas supremamente grande y por lo tanto el procesamiento llevaría mucho tiempo, aún para un supercomputador.
Con el propósito de mitigar el problema anterior; en [7] se presenta una modificación del algoritmo NW, llamado FDASA, donde se calcula únicamente la parte de mayor interés en la matriz (Fig 2), su diagonal; este algoritmo se implementó en hardware con algunas mejoras y se va a nombrar como K-band.
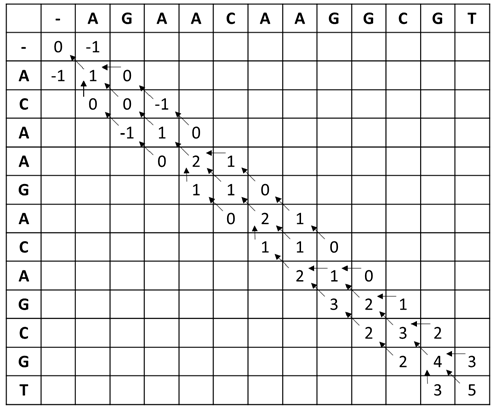
Desde la Fig. 2 se pueden observar a primera vista las ventajas del algoritmo K-band aplicado en el ejemplo presentado anteriormente. En este caso se reduce mucho la cantidad de tiempo de cálculo de la matriz de puntuación ya que sólo se procesan las 3 diagonales principales. También se puede notar que para este ejemplo, el proceso de trace-back no afecta el alineamiento de las secuencias del ejemplo pues la suma absoluta de inserciones y supresiones no es mayor a 1. Sin embargo, en la naturaleza es posible encontrar un número mayor de relación inserciones-supresiones en las secuencias.
Para enfrentar este problema, el algoritmo k-band, una modificación del algoritmo NW, realiza el cálculo únicamente de la diagonal principal y de un número K de diagonales subsecuentes a ésta dentro de la matriz, tal como es mostrado en la Fig. 3.
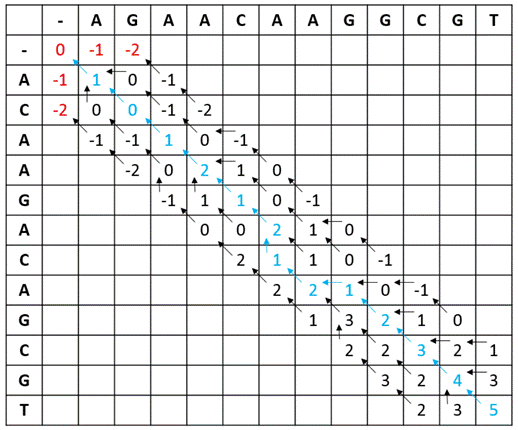
Desde la Fig. 3 se puede observar que la ampliación de la banda a cuatro diagonales desde la principal no altera el resultado para el cálculo del camino de regreso, marcado con azul, pues como se había mencionado anteriormente este ejemplo no presenta una suma absoluta de GAPs superior a 1. En contraste el aumento de la banda tiene como efecto el cálculo de otras dos diagonales cuya información es irrelevante, esto se puede notar ya que ninguna de sus celdas está dentro del camino trazado por el trace-back.
Por lo tanto, en este caso, aumentar el tamaño de la banda K resultaría en un aumento de cálculos de pesos en la matriz que no son relevantes para el alineamiento óptimo. En el caso que el alineamiento tenga una suma absoluta de GAPs mayor que la banda escogida, sí habrá una pérdida de información por lo que será necesario aumentar la banda hasta que el valor total de puntuación (score) del alineamiento deje de aumentar.
IV. DISEÑO DE LA ARQUITECTURA HARDWARE
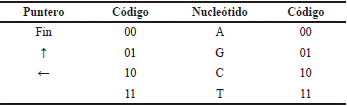
La siguiente sección describe los bloques funcionales del diseño del arreglo paralelo 1D. Las bases nitrogenadas y los punteros son codificados usando 2 bits cada uno, como se muestra en la Tabla I . Los bloques son descritos en VHDL genérico, permitiendo modificar fácilmente el número de nucleótidos de las secuencias a alinear, los valores de gap, coincidencia (match) y no coincidencia (missmatch) de las bases y el tamaño de la banda K.
1. Unidad de procesamiento
La unidad básica de cálculo (UP) es mostrada en la Fig. 4 y contiene dos comparadores (con dos y tres entradas) y tres sumadores de 8-bits sin bit de desborde (ya que los pesos son mayores al valor de gap) que permiten calcular el valor del puntaje y la dirección del puntero de una celda.
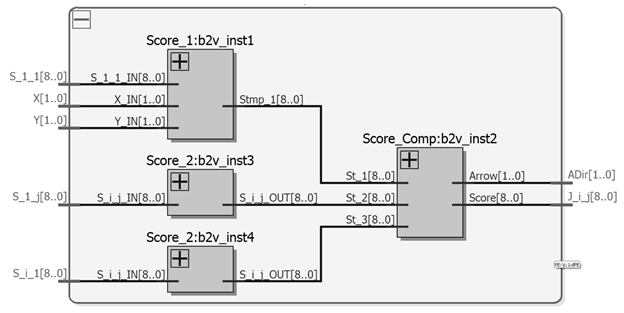
Score_1 calcula el valor de puntación sumando el valor de su entrada correspondiente al puntaje de la celda diagonal anterior F(i-1,j-i) con el valor de s(xi,xj), el cual corresponde a la ponderación de acierto o diferencia (Match - Missmatch) en la comparación de los dos nucleótidos de las secuencias de entrada.
Score_2 calcula el valor de puntuación restándole el valor predefinido del gap (d) al valor del puntaje de la celda superior.
Score_3 calcula el valor de puntuación restándole el valor predefinido del gap (d) al valor del puntaje de la celda a su izquierda.
Score Comp realiza la comparación entre los valores de los tres resultados anteriores para determinar cuál es el valor de puntaje de la celda actual y la dirección de donde proviene el mismo.
2. Módulo de procesamiento
El bloque denominado módulo de procesamiento (MP) se encarga de realizar el cálculo de los valores de puntuación de cada una de las celdas contenidas en la banda durante cada paso del algoritmo. Este bloque consta de unidades de procesamiento interconectadas y se sintetiza con diferente número de celdas dependiendo del valor de K. La Fig.5 muestra el RTL de un MP con K=2 mientras en la Fig. 6 se muestra para K=4.
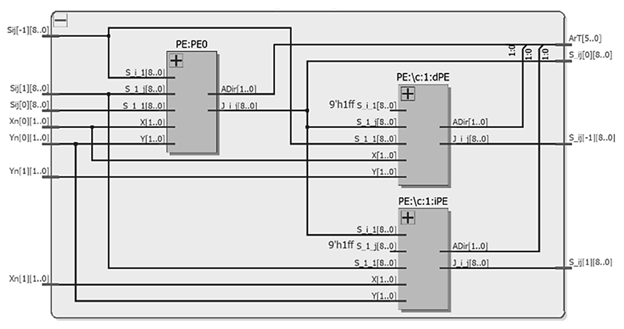
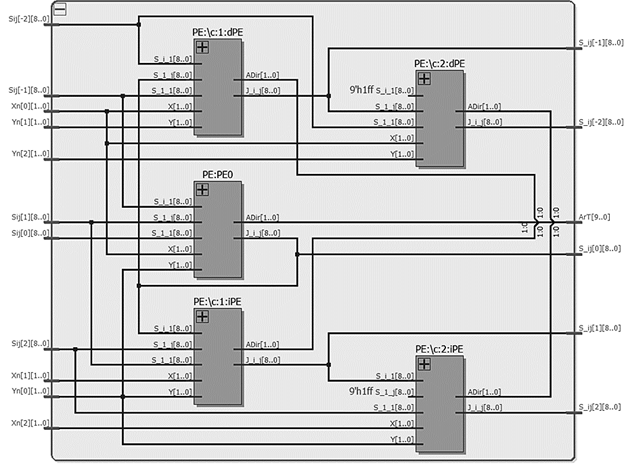
De las Fig. 5 y 6, se observa que a medida que crece el tamaño de la banda, así mismo crece el número de UPs que se necesitan en el módulo de procesamiento debido a que el número de celdas dentro de la banda será mayor. Esto hace que el sistema completo requiera más hardware en el momento de la implementación si se incrementa el valor K.
Desde la Fig. 7 se puede observar la secuencia de procesamiento de cada MP dentro de la matriz de pesos definido por el tamaño de la banda. En este caso (K=4), cada MP procesa 5 celdas básicas correspondiente a las celdas de la banda y a la diagonal principal. Por lo tanto, la banda K es un número que siempre debe ser par y el número de celdas que serán procesadas en cada elemento será igual al valor de la banda más uno (#celdasxMP = K+1).
3. Arreglo paralelo 1D
La Fig. 8 muestra el diagrama de bloques del arreglo paralelo de una dimensión para el alineamiento de dos secuencias de trece nucleótidos para K=2 y K=4. Las secuencias son nombradas como X y Y con sus respectivos índices (0..12). Las salidas de cada MP se almacenan como los valores de puntuación de sus respectivas celdas y se conectan al siguiente MP para el cálculo de los valores adyacentes.
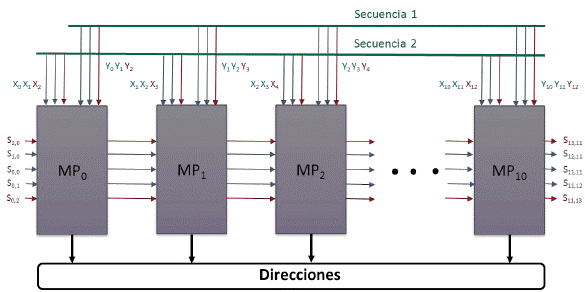
Como se puede observar en la Fig. 8, si el tamaño de la banda aumenta, el sistema se sintetiza con bloques de mayor área y el número de entradas se aumenta. En el ejemplo de la Fig. 3 se muestra el resultado de incrementar K de 2 a 4 y las dos salidas extra de cada MP resultan en dos valores de puntuación extra por fila-columna.
V. MATERIALES, PRUEBAS y RESULTADOS
Para verificar el desempeño del diseño propuesto dos secuencias se alinearon y éstas corresponden a las proteínas de transición de un ratón y una rata (NM_009407.2: Mus musculus transition protein 1 (Tnp1), mRNA. / NM_017056.2: Rattus norvegicus transition protein 1 (Tnp1), mRNA) [11,12]. En este caso, cada secuencia tiene 431 y 439 pares de bases nitrogenados, respectivamente.
Todas las simulaciones se realizaron utilizando ModelSim ALTERA STARTER EDITION 10.3c y la compilación del hardware se realizó con Quartus II v14.1.0 sobre el FPGA Cyclone EP4CGX30CF23C6. Para las pruebas se utilizaron los siguientes valores d = -1, mach = 1, missmatch = -1. El tamaño de la banda es K= [2,4,6,8,10]. Para todos los ejemplos se usó un arreglo paralelo de 10 MPs el cual fue simulado de forma cíclica hasta completar todas las bases en las secuencias. En la Tabla 2 se presentan los resultados de síntesis de hardware.
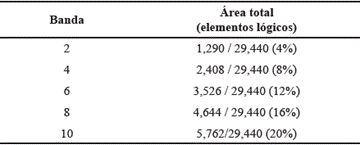
Desde la Tabla 2 es posible observar que los elementos lógicos utilizados por el diseño crecen linealmente con el aumento del valor K por lo cual se debe diseñar el arreglo con un número no muy grande de MPs para valores de banda diferentes sin llegar a afectar la frecuencia de procesamiento. A continuación se presentan los 32 primeros pares de bases nitrogenadas en la alineación, resultado de la simulación del algoritmo implementado en software, usando MATLAB y ejecutado en un PC.
X: TTCGGCAGAAAGTACCATGTCGACCAGCCGCA
Y: TT-GGCAGAAATTACAATGTCGACCAGCCGCA
Los siguientes son los últimos 32 nucleótidos de las secuencias alineadas:
X: ACA -TTTTGAAAACAAA -TAAAATTGTGAAAA
Y: ACAATTTTGAAAACAAAATAAAATTGTGAAAA
Para la simulación del algoritmo implementado en hardware, el FPGA utilizado no es el más adecuado para el diseño, sin embargo tiene la cantidad suficiente de ALUTs para sintetizar la arquitectura paralela con 10 MPs y ejecutar todo el alineamiento de manera secuencial por medio de una máquina de estados que lleva a cabo el control del sistema.
Las Fig. 9 y 10 muestran resultados parciales en el alineamiento de las secuencias para K=2 y K=10, respectivamente. Cada valor representa las puntuaciones de las celdas que ingresan al arreglo paralelo en cada iteración. Los valores en la misma columna corresponden a celdas en la misma banda.


Se puede observar en la última columna de cada simulación, que los valores de puntuación aumentan con el aumento de K. En la Tabla 3 se presenta los valores de mayor puntuación que corresponden a la última columna para cada valor de K.
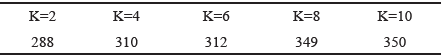
Desde la Tabla 3 se puede observar que un valor menor de K produce un menor valor en las celdas finales con respecto a la cantidad de secuencias. Esto es debido a que la banda no abarca todas las celdas del alineamiento óptimo, por lo que se presentará pérdida de información relevante. A continuación se muestran los primeros 32 pares de las secuencias alineadas de la implementación hardware pero con el valor de banda K=2:
X': TTCGGCAGAAAGTACCATGTCGACCAGCCGC-
Y': TT-GGCAGAAATTACAATGTCGACCAGCCGCA
Se puede observar con respecto al alineamiento óptimo mostrado con anterioridad, que se presenta una mutación tipo supresión adicional en el par 32 de la cadena X, lo que genera un error con respecto al alineamiento objetivo. Por lo tanto, como se puede observar en la Tabla 3, el valor de K puede ser escogido aumentando gradualmente hasta que los valores de puntuación no varíen, con ello aseguramos que no se estén agregando GAPs al alineamiento que se traduzcan en un valor de puntuación total menor.
Para verificar que el funcionamiento del sistema es independiente de la cantidad de pares de bases nitrogenadas que posean las secuencias, se realizó una segunda simulación usando dos cadenas con más de doce mil bases nitrogenadas correspondientes al RNA de la proteína de anclaje 9 para la cinasa A del caballo (Equus caballus AKAP9) [13] y el humano (Homo sapiens AKAP9) [14].
El resultado de la comparación de estas dos cadenas entregó resultados de comportamiento similar pero con valores de K mayor que las cadenas referenciadas anteriormente. En la Tabla 4 se puede observar que el valor de puntuación mayor de la última columna es pequeño para una banda de valor K=20. A medida que se va incrementando el valor de la banda, se puede notar que aumenta el valor de puntuación hasta llegar a K=80; donde se presenta el mayor valor de puntuación.
Desde estos resultados, se puede concluir que entre las dos secuencias alineadas existe una diferencia máxima de cuarenta gaps repartidos a lo largo de cada una las mismas, lo cual representa menos del uno por ciento de sus bases totales. Lo anterior es debido a que una banda de K > 80 no genera cambios en la puntuación de las últimas celdas, por lo tanto una banda que permite 40 saltos por encima o debajo de la diagonal principal contiene el camino al valor óptimo de alineamiento.

VI. CONCLUSIONES Y TRABAJO FUTURO
La implementación en hardware del alineador de secuencias de ADN presenta resultados satisfactorios con respecto al tiempo de procesamiento; el simulador ModelSim de Altera puede procesar cada iteración sobre el arreglo de elementos de procesamiento en 10ns, por lo tanto, la alineación de las 411bp de las primeras cadenas pueden ser procesadas en 0,4ms según los resultados de simulación.
Las pruebas de cambio de banda mostraron que hay una diferencia significativa entre K=2 y K=8 mientras que los incrementos siguientes de K no generaron mayor diferencia por lo que se concluye que en estas secuencias existe como máximo una diferencia de ocho inserciones-supresiones entre ambas cadenas. Así mismo, para un par de secuencias treinta veces más grande se presenta un comportamiento similar para bandas diez veces mayores.
El análisis de puntuación final en la matriz realizando cambios relativos en la banda K, permite obtener una idea muy aproximada de que tan similares son las secuencias incluso antes del alineamiento y esto se logra con la implementación del algoritmo k-band, sin la necesidad de hacer uso de todo el cálculo de la matriz, lo cual reduce considerablemente el uso de recursos de procesamiento y memoria con respecto a las implementaciones del algoritmo NW.
La posibilidad de poder sintetizar el sistema con aumentos del valor de K permite encontrar el correcto tamaño de la banda haciendo comparaciones del valor de puntuación final hasta que la banda no aumente más. De esta forma se puede encontrar el valor mínimo de la banda que abarque el alineamiento óptimo logrando que no haya pérdidas de información significativa en el alineamiento.
La implementación hardware es eficiente a pesar de estar basada en un algoritmo computacionalmente exigente como el de Needleman-Wunsch ya que permite ahorrar el cálculo de todas las puntuaciones existentes dentro de la matriz.
Desde el punto de vista del diseño digital, se llamó "genérico" a la implementación del algoritmo ya que permite sintetizar un sistema cuya banda puede variar cambiando únicamente un parámetro numérico y recompilando el código VHDL.
Una desventaja del diseño es el aumento del hardware que conlleva incrementar el parámetro K, sin embargo, dependerá del caso de análisis biológico, es decir según el número de inserciones o supresiones totales al comparar cadenas de ADN correlacionadas.
El trabajo futuro será orientado a diseñar un sistema hardware que se encargue de realizar la segunda parte del proceso de alineamiento, el traceback sobre la matriz calculada por el k-band en el diseño propuesto.

