










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkUniversitas Medica
Print version ISSN 0041-9095On-line version ISSN 2011-0839
Univ. Med. vol.59 no.2 Bogotá Apr./June 2018
https://doi.org/10.11144/javeriana.umed59-2.tran
Artículos originales
Rob(13; 15) (q10; q10) translocation: comments on a case
1Hospital Provincial General Luis G. Dávila, Tulcán, Ecuador
2Consulta externa-Centro de Salud 01, Tulcán, Ecuador
3Consulta externa-Centro de Salud 01, Tulcán, Ecuador
Introduction:
Robertsonian translocation is defined as the fusion of two non-homologous acrocentric chromosomes, with a frequency of one case per 1000 newborns.
Case report:
A 31-year-old female patient with the following gynecological and obstetrical history: gestations: 7, abortions 6, births 0, cesareans 1, children alive 1, children dead 0. Pregnancy 1: 12-year-old daughter, with no dysmorphia, from the second gestation 11 years ago to the seventh gestation occurred this year, have ended in spontaneous abortions before the first 12 weeks of gestation. With a cytogenetic study that reports Robertsonian translocation, 45, XX, t (13/15).
Conclusion
The carrier of a Robertsonian translocation between chromosomes 13;15, an event that leads to early pregnancy loss or to the birth of a neonate with multiple defects.
Keywords habitual abortion; genetic translocation; karyotype
Introducción:
La translocación robertsoniana se define como la fusión de dos cromosomas acrocéntricos no homólogos, con una frecuencia de un caso por cada 1000 recién nacidos.
Caso clínico:
Mujer de 31 años de edad, con 6 abortos. Gesta 1: hija de 12 años de edad, con ausencia de dismorfias. Terminaron en abortos espontáneos antes de las 12 primeras semanas de gestación desde la gesta 2, 11 años atrás, hasta la gesta 7, ocurrida en el año de la consulta. Con estudio citogenético que reporta 45, XX, rob(13;15) (q10;q10).
Conclusión:
El portador de una translocación robertsoniana entre los cromosomas 13;15 conduce a la pérdida precoz del embarazo o al nacimiento de un neonato con múltiples defectos.
Palabras clave aborto habitual; translocación genética; cariotipo
Introduction
One of the structural chromosomopathies is the Robertsonian translocation, defined as the fusion of two non-homologous acrocentric chromosomes (1,2,3). The frequency is one case per 1,000 newborns (4,5). It occurs with the five acrocentric chromosomes 13, 14, 15, 21 and 22 (6).
The translocation is observed in the conventional karyotype, and the number of chromosomes can be reduced if the short arm of the chromosome produced by the translocation is lost in cell division (7,8). The carriers of a Robertsonian translocation are phenotypically normal (9,10), but at the moment of wishing to have children they may present infertility, spontaneous abortions, fetal deaths and children with unbalanced chromosomal abnormalities (11,12).
Case report
The case corresponds to a 31-year-old woman referred for the first time to the gynecology area of the emergency service, due to an incomplete abortion. In conditions of asepsis and antisepsis, a manual endometrial aspiration was performed under general anesthesia, without complications.
The patient returned later to the gynecology outpatient clinic, where autoimmunity tests were performed: immunoglobulin (IgM) antiphospholipid antibody: <10 U/ml; lupus anticoagulant: 30 seconds; IgM cardiolipin: < 7 U/ml, which were all negative.
The patient was referred to the genetic counseling clinic for having a history of six miscarriages, always occurring in the first trimester of pregnancy. In this consultation an exhaustive anamnesis was made, with elaboration of the genealogical tree and physical examination of the patient. The findings were: height: 1.62 m; weight: 83.3 kg; body mass index: 32.13; blood pressure: 116/76 mmHg; heart rate: 98 beats per minute; respiratory rate: 20 per minute; temperature: 36.1 °C. Normal female phenotype, with conserved intelligence.
The patient is the first of two daughters of a couple without a history of consanguinity or inbreeding. She had an adequate growth in childhood and complete primary schooling; she has the following gynecological history: menarche at the age of eleven, regular menstrual cycles with menstruation for four days. Gestations: 7; abortions: 6; births: 0; caesarean sections: 1; living children: 1; dead children: 0.
Pregnancy 1: daughter of 12 years of age, which she gave birth by caesarean section, with absence of dysmorphisms. The next 6 pregnancies ended in spontaneous abortions before the first 12 weeks, from the second pregnancy, 11 years ago, to the seventh, occurred in the year of the consultation.
Laboratory tests reported: normal blood count and biochemistry, fasting glucose: 89 mg/dl, with thyroid stimulating hormone (TSH): 2.1 mU/l, and free T4: 1.2 ng/dl. The ultrasound scan of the uterine cavity did not show anatomical abnormalities, with normal adnexa.
After obtaining an informed consent from the patient, a 3 ml sample of peripheral blood was taken in a sterile heparinized tube to prevent coagulation and to keep the lymphocytes free (13). The culture medium was enriched with exogenous supplement, which provides the cells with favorable conditions with nutrients and growth factors. This allowed to initiate and maintain sufficient cell cycles in vitro (11,14).
T lymphocytes were stimulated with filamentous hemagglutinin (FHA) and incubated at 37 °C for 72 h. The culture was treated with colchicine, which inhibited the formation of the spindle apparatus, and incubated at 37 °C for several minutes. The harvest of 20 metaphases was made once the metaphase culture was stopped. We proceeded to perform a first centrifugation; later, we extracted the surplus, which was replaced by an equal amount of a hypotonic solution, and we carried out the second centrifugation. For the fixation, acetic acid and methanol were used, with extension on a glass slide, in addition to the staining with trypsin-giemsa. Each chromosome was analyzed under the optical microscope, considering the presence and the pair of the human chromosomes in a well-defined order The result of the proband sample corresponded to 45, XX, rob(13;15) (q10; q10) (Figure 1).
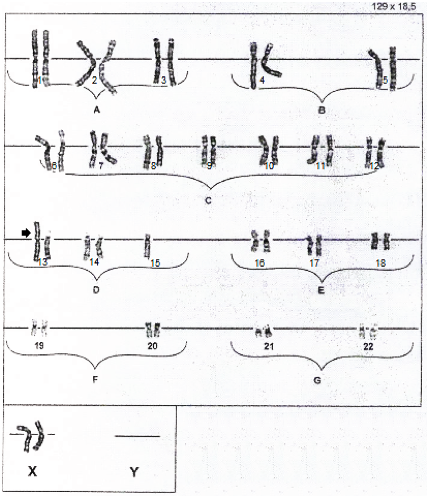
Discussion
A Robertsonian translocation occurs due to the breakage of two acrocentric chromosomes (numbers 13, 14, 15, 21 and 22) or near the centromeres, with the subsequent fusion of their long arms (14,15).
Since there is no loss or gain of important genetic material, it is considered a functionally balanced reconfiguration (15,16). The resulting balanced karyotype (II-7) has 45 chromosomes with the translocated chromosome, which is formed by the long arms of chromosomes 13 and 15.
On the other hand, the short arms of the chromosomes involved in the fusion are often lost. Apparently, these arms have no clinical importance (17,18), since they only have ribosomal RNA genes, for which there are multiple copies in the other acrocentric chromosomes (15).
Robertsonian translocations can be monocentric or pseudo-dicentric (17). In our case, the breaking point was located in the centromeric region, whose union is called centric fusion (15).
Miryounesi et al. (5) reported that one of the most common Robertsonian translocations were 13q14q and 14q21q, while among the least common chromosomal rearrangements (17,19,20), with 4.12%, rob(13;15) (q10;q10) was found, which is present in our case.
Most heterozygous of Robertsonian translocation are inherited from a carrier father of mother; while the minority are de novo, a result that has been generated in meiosis I of oogenesis (11,19). The presence of a heterozygous carrier daughter (II-7) phenotypically normal in the second generation of the genogram (Figure 2) identifies the I-5 man or the I-6 woman as possible carriers. In general, female carriers show an increased risk of transmitting the translocation to their offspring (17), a situation that should be defined by the cytogenetic study.
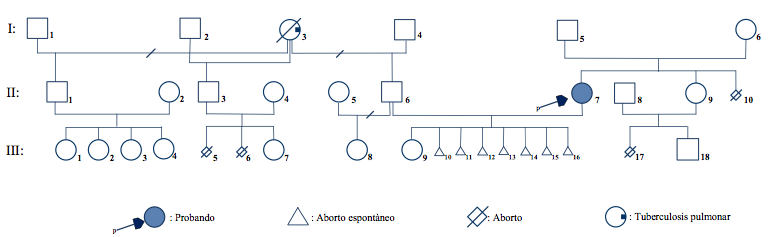
The abortions (III-10/III-16) of the II-6 II-7 couple raises the question of which of the parents carries the translocation. Thus, man II-6 has a normal karyotype, 46, XY.
In theory, the gametes that the heterozygous carrier (II-7) can produce (Figure 3) are: (1) a normal chromosomal complement, that is, a 13 and a normal 15. (2) A balanced chromosomal complement, given by the chromosome with the 13/15 translocation (15,17,21). Here the gametes are viable, and proof of this is the presence of III-9 in the third generation of the genogram, with the question of whether she is a carrier like her mother, who is phenotypically normal. (3) An unbalanced chromosome of the gamete, which has the chromosome with the 13/15 translocation plus the normal 13; this would cause the fertilized embryo to present a trisomy 13. (4) The unbalanced chromosomal complement with normal 13 and absence of a 15. (5) An unbalanced chromosomal complement with chromosome 13/15 of the translocation, and a normal chromosome 15. (6) The unbalanced chromosomal complement with normal 15 and absence of a 13 (11,15). The non-shaded gametes on the right produce non-viable offspring, events that explain the early pregnancy losses in the patient proband (II-7).
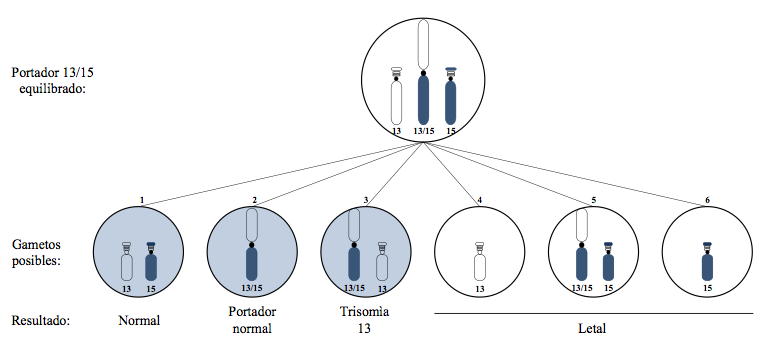
In summary, there are six possibilities of separation of the chromosomes involved from the tetrad in the formation of the gametes. For each meiosis of II-7, three of the gametes do produce viable offspring; of the three viable, one is normal, another balanced and another unbalanced with chromosome 13 translocated and 13 normal. In combination with a normal gamete, the latter can result in a child with translocational trisomy 13 (15,17,22).
Conclusion
The carrier of a Robertsonian translocation between chromosomes 13;15 has only 45 chromosomes, an event that leads to an early pregnancy loss or to the birth of a neonate with multiple defects. In theory, the chromosomal complement that can form a carrier in the gametes are three viable: one normal, another balanced, trisomy 13, and three unbalanced gametes that are incompatible with life beyond the first trimester of pregnancy.
The risk of recurrence is high in families in which a parent is a carrier of the translocation. Because of this, before offering genetic counseling, it is necessary to karyotype the parents, even including other relatives.
Referencias
1. Abdalla EM, Kholeif SF, Elshaffie RM. Homozygosity for a Robertsonian Translocation (13q;14q) in an otherwise healthy 44, xy man with a history of repeated fetal losses. Lab Medicine. 2013;44(3):254-7. [ Links ]
2. Wang B, Xia Y, Song J, Wang W, Tang Y. Case report: Potential speciation in humans involving Robertsonian translocations. Biomed Res. 2013;24(1):171-4. [ Links ]
3. Online Mendelian Inheritance in Man-OMIM [Internet]. 2016 [citado 2017 may]. Disponible en: http://www.ncbi.nlm.nih.gov/Omim/. [ Links ]
4. Song J, Li Xi, Sun Lei, Xu S, Liu N, Yao Y, et al. A family with Robertsonian translocation: A potential mechanism of speciation in humans. BioMed Central. 2016;1(2):1-7 [ Links ]
5. Miryounesi M, Diantpour M, Motevaseli E, Ghafouri-Fard S. Homozygosity for a Robertsonian translocation(13q;14q) in a phenotypically normal 44, xx female with a history of recurrent abortion and a normal pregnancy outcome. J Reprod Infertil. 2016;17(3):184-7. [ Links ]
6. Solari A. Fundamentos y aplicaciones en medicina: genética humana. 3a ed. Bogotá: Editorial Médica Panamericana; 2008. [ Links ]
7. Slovak M, Theisen A, Shaffer LG. Human chromosome nomenclature: An overview and definition of terms. En: Gersen S, Keagle M. The principles of clinical cytogenetic. 3a rev ed. New York: Springer Science; 2013. p. 34-6. [ Links ]
8. Cruz M, Bosh J. Atlas de síndromes pediátricos. Barcelona: ESPAXS; 2008. [ Links ]
9. Bacolla A, Wells R. Non-B DNA and chromosomal rearrangements. En: Lupski J, Stankiewicz P. The genomic basis of disease. New Jersey: ANSI; 2006. p. 93-4. [ Links ]
10. Lyons K. SMITH Patrones reconocibles de malformaciones humanas. 6a ed. Barcelona: Elsevier Saunders; 2008. [ Links ]
11. Strachan T, Read A. Genética humana. 3ª ed. New York: McGraw-Hill Interamericana; 2011. [ Links ]
12. Ecuador, Ministerio de Salud Pública. Diagnóstico y tratamiento del aborto espontáneo, incompleto diferido y recurrente: guía de práctica clínica. Quito: s. e.; 2013. [ Links ]
13. Lantigua A. Introducción a la genética médica. 2a ed. La Habana: Ciencias Médicas; 2011. [ Links ]
14. Luque J, Herráez A. Biología molecular e ingeniería genética: conceptos, técnicas y aplicaciones en ciencias de la salud. La Habana: Ciencias Médicas; 2001. [ Links ]
15. Turnpenny P, Ellard S. Emery´s elements of medical genetics. 13a ed. Edimburgo: Elsevier Limited; 2007. [ Links ]
16. Paz-Y-Mino C, López-Cortés A. Genética molecular y citogenética humana: fundamentos, aplicaciones e investigaciones en el Ecuador. Quito: Yachay; 2014. [ Links ]
17. Nussbaum R, Mclnnes R, Willard H. Thompson-Thompson Genética médica. 7a ed. Barcelona: Elsevier Masson; 2009. [ Links ]
18. Yip MY. Uniparental disomy in Robertsonian translocations: strategies for uniparental disomy testing. Transl Pediatr. 2014;3(2):98-107. [ Links ]
19. Hasanzadeh-Nazar M, Baghbani F, Namazi I, Mirzaee S. Robertsonian translocation between chromosomes (nº 21/14) in relation to the history of spontaneous abortion in a family. J Reprod Med. 2014;12(8):581-5. [ Links ]
20. Zhao W-W, Menghua W, Chen F, Jiang S, Su H, Liang Jianfen, et al. Robertsonian traslocations: an overview of 872 Robertsonian translocation identified in a Diagnostic laboratory in China. PlosOne. 2016;10(5):872-87. [ Links ]
21. Xu SQ, Tang DL, Fang K, Xia YZ, Song JP, Wang WP, et al. Analysis of meiotic segregation patterns and interchromosomal effects in sperm from a Robertsonian translocation family. Biomed Res. 2014;25(2):233-9. [ Links ]
22. Kochhar PK, Ghosh P. Reproductive outcome of couples with recurrent miscarriage and balanced chromosomal abnormalities. J Obstet Gynaecol Res. 2013;39(1):113-20. [ Links ]
Received: May 31, 2017; Accepted: October 27, 2017
 Esta obra está bajo una Licencia Creative Commons Atribución 4.0 Internacional.
Esta obra está bajo una Licencia Creative Commons Atribución 4.0 Internacional.
