











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroducción
Entre las cromosomopatías estructurales está la translocación robertsoniana, definida como la fusión de dos cromosomas acrocéntricos no homólogos (1,2,3). La frecuencia es de un caso por cada 1000 recién nacidos (4,5). Ocurre con los cinco cromosomas acrocéntricos 13, 14, 15, 21 y 22 (6).
La translocación es observada en el cariotipo convencional y puede reducirse el número de cromosomas si el brazo corto del cromosoma producto de la translocación se pierde en la división celular (7,8). Los portadores de una translocación robertsoniana son fenotípicamente normales (9,10); pero en el momento de desear hijos pueden presentar infertilidad, abortos espontáneos, muertes fetales e hijos con anomalías cromosómicas no balanceadas (11,12).
Caso clínico
El caso corresponde a una mujer de 31 años de edad referida por primera vez al área de ginecología, en el servicio de emergencia, por un aborto incompleto. Bajo normas de asepsia y antisepsia, con anestesia general, se le realizó una aspiración manual endometrial, sin complicaciones.
Posteriormente, la paciente retornó a la consulta externa de ginecología, donde se le realizaron exámenes de autoinmunidad: anticuerpo antifosfolípido de inmunoglobulina (IgM): < 10 U/ml; anticoagulante lúpico: 30 segundos; cardiolipina IgM: < 7 U/ml, que resultaron todos negativos.
La paciente fue remitida a la consulta de asesoramiento genético por tener antecedente de seis abortos, ocurridos siempre en el primer trimestre del embarazo. En esta consulta se realizó una exhaustiva anamnesis, elaboración de árbol genealógico y examen físico de la paciente. Los hallazgos fueron: talla: 1,62 cm; peso: 83,3 kg; índice de masa corporal: 32,13; tensión arterial: 116/76 mmHg; frecuencia cardiaca: 98 latidos por minuto; frecuencia respiratoria: 20 por minuto; temperatura: 36,1 °C. Fenotipo femenino normal, con inteligencia conservada.
Es la primera de dos hijas de una pareja sin historia de consanguinidad o endogamia. Tuvo crecimiento adecuado en la niñez e instrucción primaria completa. Y cuenta con los siguientes antecedentes ginecobstétricos: menarquia a los once años, ciclos menstruales regulares, con menstruación por cuatro días. Gestas: 7; abortos: 6; partos: 0; cesáreas: 1; hijos vivos: 1; hijos muertos: 0.
Gesta 1: hija de 12 años de edad, quien fue obtenida por cesárea, con ausencia de dismorfias. Terminaron en abortos espontáneos antes de las 12 primeras semanas de gestación las siguientes 6 gestas. Desde la segunda, 11 años atrás, hasta la séptima, ocurrida en el año de la consulta.
Los estudios de laboratorio reportaron: hemograma y bioquímica normal, glucosa en ayunas: 89 mg/dl, con hormona estimulante de la tiroides (TSH): 2,1 mU/l, y T4L: 1,2 ng/dl. El rastreo ecográfico de la cavidad uterina no evidenció anomalías anatómicas, con anexos normales.
Después de obtener el consentimiento informado de la paciente, se procedió a tomar una muestra de 3 ml de sangre periférica, en un tubo estéril heparinizado, de tal manera que se impidiera la coagulación y los linfocitos se mantuvieran libres (13). El medio de cultivo fue enriquecido con suplemento exógeno, que proporciona a las células condiciones favorables con nutrientes y factores de crecimiento. Ello permitió iniciar y mantener suficientes ciclos celulares in vitro (11,14).
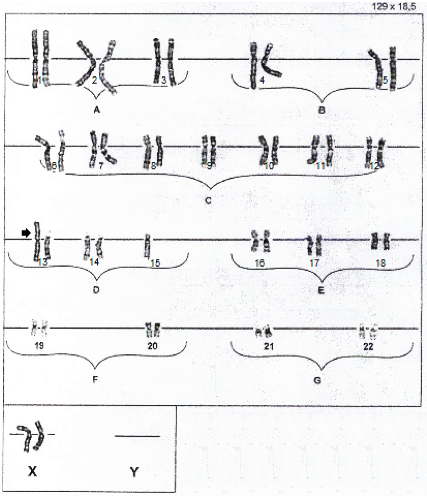
Los linfocitos T se estimularon con fibrohemaglutinina (FHA) y se incubaron a 37 ºC durante 72 h. El cultivo estuvo tratado con colchicina, que inhibió la formación del huso cromático, e incubamos a 37 ºC durante varios minutos. La cosecha de 20 metafases se realizó una vez detenido el cultivo en metafase. Procedimos a realizar una primera centrifugación; posteriormente, extrajimos el sobrante, que fue sustituido por igual cantidad de una solución hipotónica, y llevamos a cabo la segunda centrifugación. Para la fijación se utilizó ácido acético y metanol, con extensión sobre una lámina portaobjeto, además de la coloración con tripsina-giemsa. En la observación bajo el microscopio óptico, se analizó cada cromosoma, considerando la presencia y la pareja de los cromosomas humanos en orden bien definido. El resultado de la muestra del probando correspondió a 45, XX, rob(13;15) (q10;q10) (figura 1).
Discusión
Una translocación robertsoniana ocurre por la rotura de dos cromosomas acrocéntricos (números 13, 14, 15, 21 y 22) o cerca de los centrómeros, con la posterior fusión de sus brazos largos (14,15).
Como no hay pérdida ni ganancia de material genético importante, se considera una reconfiguración funcionalmente equilibrada (15,16). El cariotipo equilibrado resultante (II-7) tiene 45 cromosomas con el cromosoma translocado, que está formado por los brazos largos de los cromosomas 13 y 15.
Por otro lado, a menudo, se pierden los brazos cortos de los cromosomas involucrados en la fusión. Estos brazos, al parecer, no tienen importancia clínica (17,18), puesto que solo tienen genes de ARN ribosómico, para el cual existen múltiples copias en los demás cromosomas acrocéntricos (15).
Las translocaciones robertsonianas pueden ser monocéntricas o seudodicéntricas (17). En nuestro caso, la localización del punto de ruptura fue en la región centromérica, cuya unión se denomina fusión céntrica (15).
Miryounesi y colaboradores (5) publicaron que entre las translocaciones robertsonianas más frecuentes estuvieron la 13q14q y la 14q21q; mientras que entre el reordenamiento cromosómico menos común (17,19,20), con el 4,12 %, se encuentra la rob(13;15) (q10;q10), presente en nuestro caso.
La mayoría de los heterocigotos de translocación robertsoniana se heredan a partir de un padre o madre portador; mientras que la minoría son de novo, resultado que se ha generado en la meiosis I de la ovogénesis (11,19). La presencia de una hija heterocigota portadora (II-7) fenotípicamente normal en la segunda generación del familiograma (figura 2) identifica al hombre I-5 o a la mujer I-6 como posibles portadores. En general, las mujeres portadoras muestran un riesgo incrementado de transmitir la translocación a su descendencia (17), situación que se debería definir con el estudio citogenético.
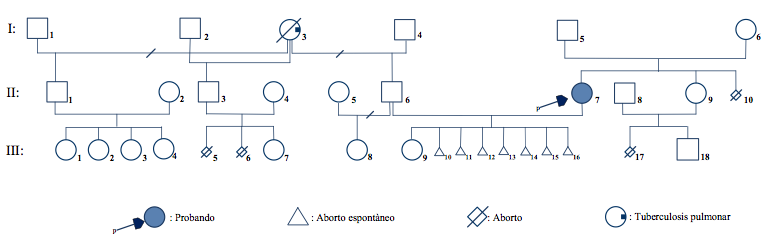
Los abortos (III-10/III-16) de la pareja II-6 y II-7 crean la interrogante de conocer cuál de los padres es el portador de la translocación. Así, el hombre II-6 presenta un cariotipo normal, 46,XY.
En teoría, los gametos que la heterocigota portadora (II-7) puede producir (figura 3) son: 1) un complemento cromosómico normal, es decir, un 13 y un 15 normal. 2) Un complemento cromosómico equilibrado, dado por el cromosoma con la translocación 13/15 (15,17,21). Aquí los gametos son viables, y muestra de ello es la presencia de III-9 en la tercera generación del familiograma, con la interrogante de si es portadora igual que su madre, fenotípicamente normal. 3) Un cromosoma desequilibrado del gameto, que posea el cromosoma con la translocación 13/15 más el 13 normal; esto desencadenaría que el embrión fertilizado presente una trisomía 13. 4) El complemento cromosómico desequilibrado con el 13 normal y ausencia de un 15. 5) Un complemento cromosómico desequilibrado con el cromosoma 13/15 de la translocación y un cromosoma 15 normal. 6) El complemento cromosómico desequilibrado con el 15 normal y ausencia de un 13 (11,15). Los gametos no sombreados a la derecha producen descendencia no viable, acontecimientos que explican en la paciente probando (II-7) las pérdidas precoces de los embarazos.
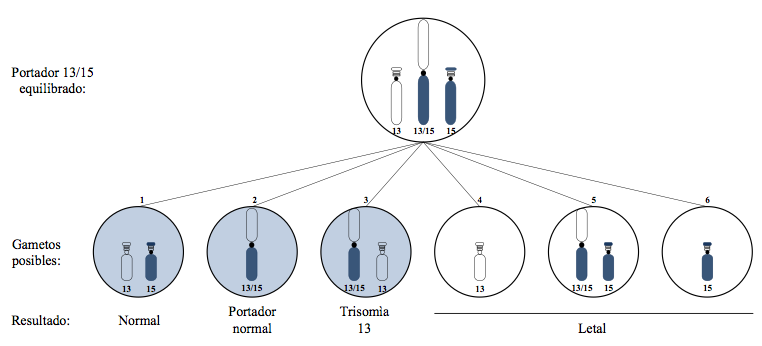
En resumen, son seis las posibilidades de separación de los cromosomas involucrados de la tétrada en la formación de los gametos. Por cada meiosis de II-7, tres de los gametos no dan lugar a descendencia viable; de los tres viables, uno es normal, otro equilibrado y otro desequilibrado con el cromosoma 13 translocado y el 13 normal. En combinación con un gameto normal, este último puede dar lugar a un niño(a) con trisomía 13 por translocación (15,17,22).
Conclusión
El portador de una translocación robertsoniana entre los cromosomas 13;15 tiene solo 45 cromosomas, acontecimiento que conduce a la pérdida precoz del embarazo o al nacimiento de un neonato con múltiples defectos. En teoría, el complemento cromosómico que puede formar un portador en los gametos son tres viables: uno normal, otro equilibrado, la trisomía 13 y tres gametos desequilibrados que son incompatibles con la vida más allá del primer trimestre de embarazo.
El riesgo de recurrencia es elevado en familias en las que un progenitor es portador de la translocación. Por lo anterior, es necesario cariotipar a los progenitores, sin dejar de lado a otros familiares, antes de ofrecer asesoramiento genético.

