











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La hiperostosis cortical infantil la describieron por primera vez, en 1945, el profesor radiólogo pediatra John Caffey y W. A. Silverman, que la definieron como una enfermedad infantil hereditaria, con una edad media de inicio a partir de las nueve semanas de vida y establecieron los sitios de hiperostosis (1).
La enfermedad de Caffey, también llamada hiperostosis cortical infantil, se caracteriza por la presencia de un episodio en la infancia con neoformación subperióstica que involucra las diáfisis de los huesos largos, el maxilar inferior y las clavículas. El cambio del hueso inicia antes de los 5 meses de edad y usualmente se resuelve antes de los 2 años (2,3).
En Estados Unidos tiene una prevalencia estimada de 3 por cada 1000 lactantes de 6 meses o menos, sin predilección por el sexo o el origen étnico (1). En Colombia, al parecer, no se ha informado de ningún caso.
Las más de mil mutaciones que se han encontrado en el mismo gen pueden dar lugar a diferentes trastornos clínicos; y entre las colagenopatías se incluyen la osteogénesis imperfecta, el síndrome de Ehlers-Danlos y la enfermedad de Caffey (4).
La mutación asociada con la enfermedad de Caffey se produce por una transición 3040C > T de un solo nucleótido en el exón 41 del gen COL1A1, de arginina por cisteína. El locus está en el cromosoma 17q21.33, con patrón de herencia autosómico dominante, de expresividad variable y heterogeneidad alélica (5). Además, se han descrito hallazgos que se correlacionan con el síndrome de Ehlers-Danlos tipo III (6).
Bennett et al. y más tarde Lecolier et al. describieron una forma letal prenatal de la enfermedad de Caffey con distintas lesiones óseas que son reconocidas ecográficamente en promedio a las 27 semanas de gestación (7,8).
En cuanto a las formas esporádicas de hiperostosis cortical infantil, se atribuyen a la administración de prostaglandinas (PGE1 y PGE2) utilizadas para tratar lesiones cardiacas cianozantes dependientes del conducto arterial, cuando reciben administración profiláctica de PGE1 para prevenir su cierre. Entre otras causas, también se incluyen el trauma, la hipervitaminosis A, la hiperfosfatemia e infecciones (9).
El objetivo del presente reporte de caso familiar consistió en describir los hallazgos clínico-radiológicos de varios pacientes de la misma familia con hiperostosis cortical, elaboración del árbol genealógico, análisis radiológico de algunos de los afectados y revisión de la literatura.
El protocolo de investigación fue enviado al comité de ética para su comentario, revisión y aprobación, lo que permitió el registro de datos del caso índice y sus familiares con la publicación de los resultados, posteriormente.
Caso clínico 1
El primer caso correspondió a un bebé recién nacido de 16 horas de vida, con madre de 25 años de edad y padre de 26 años, no consanguíneos. Era resultado de una tercera gestación, con nueve controles prenatales (tres privados) desde el inicio y siete ecografías normales. La madre no tuvo antecedentes de hipervitaminosis A o hiperfosfatemia durante el embarazo. El estudio de toxoplasma, rubeola, citomegalovirus y herpes virus fue negativo, y no hubo infecciones de las vías urinarias.
El recién nacido vivo era producto del tercer embarazo de 38,2 semanas, según la fecha de la última menstruación. Se obtuvo vía cesárea por pelviano, con líquido amniótico claro, con antropometría que correspondió a un recién nacido a término, grande para la edad gestacional, con un peso de 3575 g, talla de 51 cm y perímetro cefálico de 36 cm, con Apgar de 8 al minuto y a los 5 min.
Antes de las 24 h de vida, el recién nacido fue interconsultado con el servicio de asesoramiento genético, por aparente acortamiento y arqueamiento distal del antebrazo izquierdo y miembros inferiores.
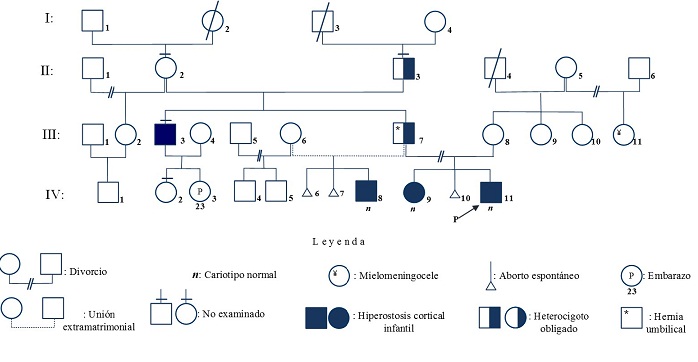
En esta consulta se realizó una exhaustiva anamnesis, elaboración de árbol genealógico y examen físico del paciente. Se recabó un episodio de inflamación e inclinación de la pierna derecha en el tío paterno durante la infancia, no así en el padre del paciente (figura 1).
En el examen físico inicial el recién nacido presentó frecuencia cardiaca de 140 latidos por minuto, frecuencia respiratoria de 57 respiraciones por minuto y saturación del 90 %. Hemodinámicamente, estaba estable y bien hidratado.
Fenotípicamente se observó:
Cráneo: occipucio, relativamente plano; con cabello fino, flexible y escaso, así como fontanela anterior normotensa.
Facies: redonda, frente inclinada con leve hirsutismo, mejillas gruesas.
Ojos: fisuras palpebrales cortas.
Nariz: pequeña con fosas nasales levemente antevertidas, y raíz nasal amplia.
Boca: maxilar inferior relativamente pequeño, comisuras labiales con dirección hacia abajo.
Orejas: pabellones auriculares de normo implantación, hélice grueso y lóbulos auriculares ausentes.
Tórax: simétrico con murmullo vesicular conservado y ruidos cardiacos rítmicos.
Abdomen: cordón umbilical con 2 arterias y una vena.
Región inguinal: genitales masculinos, testículos en bolsas escrotales.
Además, al examinarlo, en la extremidad superior izquierda se observó deformidad angular anterior de su antebrazo, con contractura en ligera flexión del codo y desviación cubital de la mano, con llanto al tacto, sin crepitación a la palpación. No constatamos equimosis ni datos de sangrado o heridas. Mientras que los miembros inferiores evidenciaron resistencia importante a la extensión de las rodillas, con aumento de volumen, moderado eritema y arqueamiento distal de las piernas e inversión de los pies. El examen neurológico fue normal.
De acuerdo con los exámenes de laboratorio realizados, había leucocitosis con fosfatasa alcalina de 472 U/L (50-120 U/L). El tamizaje metabólico neonatal reportó: 17 OH de progesterona: 9,18 nmol/L (< 27 nmol/L); hormona estimulante de la tiroides total: 1,31 mUI/L (< 20 mUI/L); galactosa: negativo; prueba de fenilcetonuria: 78,79 µmol/L (< 182 µmol/L), normal, y el análisis citogenético convencional, con técnica de bandas GTG y conteo de 20 metafases demostró cariotipo normal masculino, 46, XY.
La radiografía simple neonatal de miembros superiores e inferiores (figuras 2a y 2b) evidenció la periostitis e hiperostosis cortical con curvatura anterior del radio izquierdo, asociado con un importante engrosamiento cortical en la diáfisis de las tibias derecha e izquierda, e inflamación del tejido blando circundante. La radiografía simple de cráneo no evidenció microfracturas o engrosamiento cortical de la bóveda craneal. La ecografía abdominal mostró riñones de características conservadas, así como uréteres y vejiga sin alteraciones.
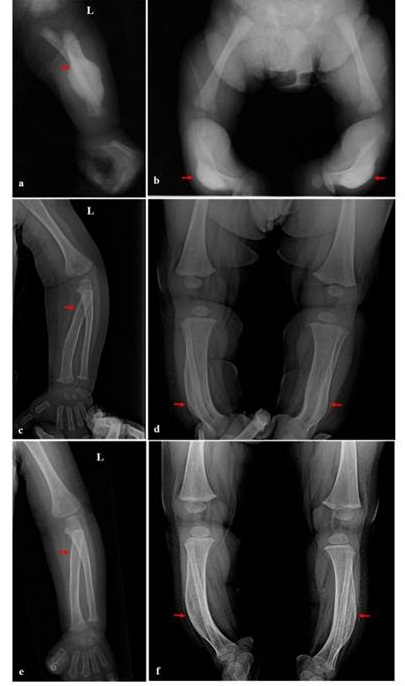
Figura 2 Elevación perióstica y engrosamiento cortical de la diáfisis. Radiografías anterolaterales simples del probando (individuo IV-11) al nacimiento. a) Radio izquierdo, b) tibia derecha e izquierda. A los tres meses; c) radio izquierdo, que con una marcada disminución perióstica; d) tibias que evidencian una moderada reducción cortical de la diáfisis. A los ocho meses, e) y f) aumento del espacio medular propio de la fase de remodelación en el radio izquierdo y tibias, respectivamente.
El paciente fue valorado por el servicio de oftalmología, que reportó fondo de ojo normal. El examen audiológico, valorado a través de potenciales evocados auditivos de tronco cerebral fue normal.
Una vez obtenidos los resultados de los exámenes iniciales realizados y valoraciones médicas, el bebé fue enviado a casa, en condición estable, con fórmula de antipirético y antinflamatorio. No fue considerado pertinente el uso de corticoide.
La evaluación radiográfica de seguimiento en la consulta externa de los tres meses de edad (figuras 2c y 2d) mostró una disminución de la hiperostosis cortical. La porción proximal de la diáfisis radial izquierda evidenció un ligero engrosamiento interno de la corteza. Las tibias derecha e izquierda exhibieron una marcada disminución perióstica e inflamación del tejido blando circundante. A los ocho meses de edad (figuras 2e y 2f) hubo un aumento del espacio medular radial izquierdo y tibial bilateral, respectivamente.
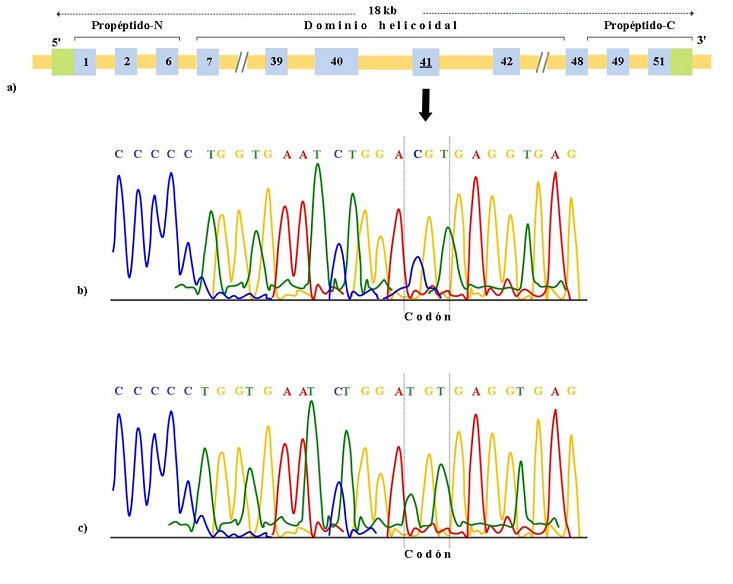
Los exámenes posteriores de laboratorio realizados a los tres meses indicaron: leucocitos de 7,360 con fosfatasa alcalina de 339,1 U/L (50-120 U/L), con disminución respecto a los valores iniciales. Entre tanto, en las pruebas moleculares del gen COL1A1 fue necesaria la amplificación de sondas dependiente de ligandos múltiples, usando un kit comercial (Invitrogen, Carlsbad, Estados Unidos) bajo las recomendaciones del proveedor o el protocolo de RT-PCR cuantitativa y la secuenciación del gen por el método de Sanger para la detección de mutaciones puntuales (figuras 3a y 3b).
En el momento de la última evaluación clínica, el paciente tenía once meses de vida, y en el examen físico presentó arcos de movimientos de las extremidades normales con ausencia de llanto.
Caso clínico 2
El caso 2 corresponde a una niña de siete años y siete meses de edad, nacida por parto eutócico a las 38 semanas de gestación, a término para la edad gestacional, con un peso de 2950 g, talla 48 cm y circunferencia de la cabeza de 33 cm. El Apgar al primer minuto fue de 7, y a los cinco minutos, de 8, con alta a los dos días.
La madre de la niña refirió que la paciente presentó varias remisiones y exacerbaciones de hiperostosis durante los primeros años de vida. Con cuatro meses de edad, presentó por aproximadamente dos meses, dolor y edema de la extremidad inferior derecha, deformidad angular anterior de pierna y contractura con ligera limitación para la flexión de la rodilla e inversión leve de pie homolateral. La hipeostosis cortical fue comprobada con radiografía del miembro inferior (no presentado). Fue tratada con antinflamatorios, y después estuvo completamente asintomática. Presentó un nuevo episodio de dolor y edema a los seis años, en la misma región, pero de la pierna izquierda. Estaba afebril. En el último año no ha presentado episodios de recurrencia. En la última evaluación clínica, a los siete años y siete meses de edad, estaba asintomática y había retomado las actividades cotidianas.
La niña fue llevada a consulta de asesoramiento genético, por una historia familiar de hiperostosis cortical infantil. En esta consulta se realizó una exhaustiva anamnesis, se le elaboró el árbol genealógico y se le realizó un examen fenotípico de la paciente. Los hallazgos fueron: talla de 127 cm, peso de 33,9 kg,(antropometría en el percentil 50, según las curvas de crecimiento del Ministerio de Salud Pública, formulario 028 A2/09); tensión arterial: 95/60 mmHg y frecuencia cardiaca de 70 latidos por minuto.
En el examen físico presentó:
Fenotipo femenino, con inteligencia normal.
Piel: lisa, suave e hiperextensible.
Cráneo: cabello fino y flexible.
Facies: frente alta, amplia con persistencia de cicatriz atrófica.
Ojos: fisuras palpebrales cortas, cejas ligeramente arqueadas y anchas, párpados gruesos y edematosos, córneas blancas.
Nariz: fina, con filtro nasal corto, pliegue nasolabial profundo
Boca: labios gruesos, apiñamiento de piezas dentarias, paladar alto y estrecho, lengua con ligera hiperextensibilidad.
Orejas: hiperextensibilidad de pabellones auriculares, hélice gruesa
Cuello: ligeramente corto.
Tórax: simétrico con murmullo vesicular conservado, ruidos cardiacos rítmicos.
Abdomen: moderada hiperelasticidad de la piel abdominal, sin evidencia de hernias.
Región inguinal: genitales femeninos.
Extremidades superiores: hipermovilidad de articulaciones pequeñas, no evidenció cicatrices en los codos.
Extremidades inferiores: máculas de hemosiderina múltiples difusas localizadas en la cara anterior y lateral de las piernas, hiperextensibilidad marcada entre dedos de los pies, presencia de arco plantar disminuido y deambulación conservada (figuras 4a-4d).
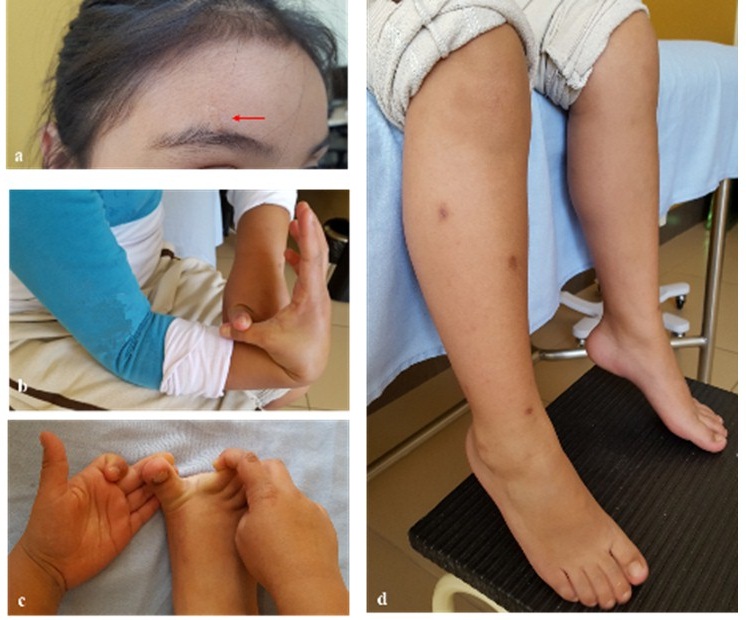
Figura 4 Niña de 7 años (paciente IV-9), que exhibe piel lisa, suave de textura aterciopelada e hipermovilidad de las articulaciones pequeñas.a) Cicatriz atrófica en la frente. b) Hipermovilidad articular; el pulgar derecho alcanza el antebrazo homolateral. c) Hiperextensibilidad entre el primer el segundo dedo del pie derecho. d) Manchas de hemosiderina en las piernas.
Los exámenes de laboratorio realizados indicaron: hemograma y bioquímica normal. Con estudio molecular que evidenció la mutación en R836C. Las radiografías actuales de tibias no evidenciaron signos de inflamación y se había eliminado el exceso de hueso periférico (no presentado). Las epífisis y los cartílagos de crecimiento fueron radiológicamente normales.
Caso clínico 3
Este caso correspondió a un lactante menor, masculino de un mes y tres días de vida. El servicio de pediatría remitió al paciente por primera vez a la consulta de asesoramiento genético por historia familiar de enfermedad ósea. En esta consulta se realizó una exhaustiva anamnesis y elaboración de su árbol genealógico (figura 1). Ello evidenció otro caso de hiperostosis cortical infantil en la localidad, del mismo padre heterocigoto obligado (III-7) de los casos anteriores descritos.
El bebé era producto de una madre de 31 años y un padre de 26 años, no consanguíneos. Era la quinta gestación. En el embarazo tuvo siete controles prenatales desde el inicio y cuatro ecografias normales. La madre no tenía antecedentes de hipervitaminosis A o hiperfosfatemia durante el embarazo. El estudio de toxoplasma, rubeola, citomegalovirus y herpes virus fue negativo, aun cuando hubo un episodio de infección de las vías urinarias, en el segundo trimestre, con tratamiento.
El bebé nació por parto eutócico a las 40 semanas de gestación, con una antropometría que correspondió a un recién nacido a término, adecuado para la edad gestacional, con peso de 3405 g, talla de 49 cm y perímetro cefálico de 34,5 cm. El Apgar fue de 9 al minuto, y a los cinco minutos.
En el examen físico presentó talla de 56,7 cm, peso de 4,7 kg, perímetro cefálico de 38,3 cm, frecuencia cardiaca de 138 latidos por minuto, frecuencia respiratoria de 54 respiraciones por minuto, saturando al 92 %.
Fenotipo masculino, irritable con llanto a la manipulación y al movimiento de extremidades inferiores. Se observó una ligera deformidad angular anterior de pantorrillas, con contractura ligera en flexión de las rodillas. No evidenciamos equimosis ni datos de sangrado o heridas (no presentado). Ausencia de dismorfias, con examen neurológico conservado.
En las radiografías de cráneo, extremidades superiores e inferiores se evidenció hiperostosis cortical de tibias e inflamación del tejido blando circundante. Los exámenes de laboratorio realizados reportaron: leucocitosis de 12.800, con fosfatasa alcalina de 1755 U/L (75-316 U/L).
En la ecografía abdominal reportó riñones de características conservadas, así como uréteres y vejiga sin alteraciones. El análisis citogenético convencional, con técnica de bandas GTG y conteo de 20 metafases, mostró un cariotipo normal masculino, 46, XY.
El paciente fue valorado por el servicio de oftalmología, que reportó fondo de ojo normal. El examen audiológico, valorado a través de potenciales evocados auditivos de tronco cerebral, fue normal. Fue iniciado manejo con antinflamatorio y analgésico, enviado a casa en condición estable y evaluación periódica en la consulta externa.
Al interpretar la genealogía familiar, se definió que los pacientes (III-3, IV-8, IV-9 y IV-11) presentaron un genotipo heterocigoto con expresión de hiperostosis, con predominio de tibias en cuatro, radio en uno, ninguno presentó en maxilar inferior, clavículas, costillas, húmero y radio (tabla 1). Se obtuvo la versión oral de III-7 respecto a un episodio de hiperostosis cortical infantil en III-3. En la edad preescolar, presentó signos clínicos de hiperlaxitud de las articulaciones, hiperelasticidad de la piel y mala cicatrización de las heridas. Tiene una niña (IV-2) aparentemente normal.
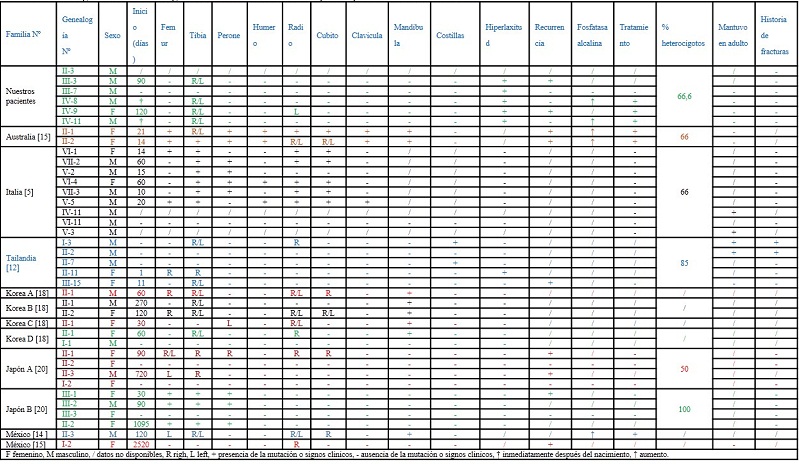
La hiperostosis cortical estaba presente en cuatro (66,6 %) heterocigotos (III-3, IV-8, IV-9 y IV-11) miembros de la familia y dos (33,3 %) eran portadores obligados (II-3, III-7), que no desarrollaron la enfermedad. De estos últimos, solo en uno (III-7) al examen físico se constató ligera hiperlaxitud articular e hiperelasticidad de la piel. Con antecedente de hernia umbilical hace dos años.
La hiperostosis cortical infantil inició entre el nacimiento y los 120 días de vida. Los individuos III-3 y III-7 tienen 30 y 26 años de edad, respectivamente. Sin antecedente de fracturas en los individuos afectados de la familia del caso índice en las cuatro generaciones. Los tres familiares del caso índice fueron examinados y evaluados utilizando radiografía simple de huesos largos y cráneo de los siete individuos vivos. Tres de los siete no fueron examinados (tabla 1).
Discusión
Los tejidos conectivos, incluido el esqueleto, están constituidos por colágeno, proteoglicanos, proteínas no colágenas, que forman la matriz extracelular. El colágeno constituye el principal componente de la matriz extracelular en el hueso, con aproximadamente el 90 % de la matriz ósea y específicamente fibras de colágeno tipo I (10,11).
La enfermedad de Caffey involucra uno o múltiples huesos, como el maxilar inferior, las clavículas, las costillas y los huesos largos, principalmente. En estos últimos la lesión primaria ósea está localizada en la diáfisis, lo que da como resultado huesos con bordes irregulares, además de la afectación de tejido conectivo contiguo. La forma infantil de la enfermedad usualmente es autolimitada, con regresión espontánea por dos años (5,12).
El caso índice (IV-11) presentó un episodio espontáneo con formación de nuevo hueso subperióstico que involucró la diáfisis de los huesos largos (tibias y radio izquierdo), con inicio antes de los primeros cinco meses de vida. Estos cambios se exhibieron en las radiografías simples; los huesos involucrados también presentaron signos de inflamación (6,13).
De acuerdo con lo observado en relación con el aumento de la formación de hueso, IV-8 y IV-11 en los hallazgos de laboratorio incluyeron elevación de la fosfatasa alcalina, la velocidad de sedimentación globular y leucocitosis. Estos indican una respuesta inflamatoria (14,15,16). En tanto que las radiografías de cráneo no contenían huesos wormianos, como ocurre en los pacientes con osteogénesis imperfecta (17). Aunque se ha informado de la resolución de síntomas, gracias al uso de la indometacina y terapia con glucocorticoides (10,16,18), solo se prescribió a IV-8, IV-11 paracetamol a una dosis de 10 mg/kg/día.
Hasta el momento, IV-9 ha experimentado dos episodios (a los cuatro meses y seis años) de remisión y exacerbación en la niñez. El diagnóstico de hiperostosis fue retrospectivo, puesto que ella no conservó las radiografías o fotografías de los episodios que sustentaran la hipótesis diagnóstica; sin embargo, por el patrón de herencia se desprende la alta sospecha clínica (19). Las radiografías actuales de tibias evidenciaron ligera expansión de la cavidad medular descrita por Glorieux (10), en la remodelación.
Los episodios recurrentes son poco frecuentes y existen escasos informes sobre secuelas adversas de la hiperostosis en individuos afectados (20). La historia natural de la enfermedad ha demostrado que los casos son autolimitados (14). En el examen físico de IV-9 se evidenció hiperlaxitud de articulaciones pequeñas, piel suave elástica y cicatrices atróficas que se asemejan a los hallazgos clínicos de una forma leve del síndrome de Ehlers-Danlos tipo III. Los defectos estructurales en la cadena de colágeno tipo 1 podrían explicar las manifestaciones clínicas en la piel y las articulaciones (5,6,21).
Gensure et al. (6) identificaron hipermovilidad articular y hernias inguinales en los tres miembros de una familia canadiense con mutación R836C, similar a nuestro estudio, puesto que III-7 fue intervenido quirúrgicamente hace dos años por una hernia umbilical.
La falta de signos clínicos en el padre (III-7) podría explicarse por la penetrancia reducida, considerando que tenga una mutación en un alelo y que el otro alelo (o una mutación en otro locus) tenga una mutación que atenúe la hiperostosis por interacción entre alelos en el padre, o si fuera en otro locus un efecto epistático podría haber atenuado el fenotipo en el padre (17).
En la hiperostosis cortical infantil, se habla de penetrancia completa cuando la mutación se expresa en el 100 % de los individuos heterocigotos (22); sin embargo, en nuestro estudio solo se encontró el 66 % de heterocigotos que tuvieron un episodio de hiperostosis cortical versus el 79 % de los individuos reportados por Gensure et al. (6).
La hiperostosis cortical infantil con herencia autosómica dominante se ha descrito con expresividad variable y heterogeneidad alélica. En las radiografías de huesos largos de los casos (IV-8 y IV-11) se observaron diferencias en el grado de hiperostosis y huesos involucrados, similar a lo descrito por Kitaoka et al. (20).
Gensure et al. (6) y Couper et al. (15) describieron la expresividad variable en individuos heterocigotos para la mutación en R836C, al considerar el número de episodios recurrentes, las manifestaciones clínicas variadas y las áreas óseas involucradas, equivalente con lo observado en nuestro estudio.
La historia clínica detallada de los individuos de la familia no reveló evento alguno precipitante que no sea la herencia autosómica dominante, con inicio del cuadro clínico antes de los cinco meses de edad (6). Ninguno de los niños (IV-8, IV-9 y IV-11) recibió prostaglandinas. El curso clínico del episodio hiperostósico fue prolongado pero resuelto completamente. A mediados de la infancia de IV-9, los huesos largos hiperostósicos se han remodelado exitosamente y con ausencia de afectación de la fisis de crecimiento (14).
Estamos en desacuerdo respecto a la persistencia de signos clínicos y radiológicos en la edad adulta. Son pocos los individuos con mutación probada en COL1A1 a largo plazo, con deformidad persistente de huesos largos. En todos nuestros individuos adultos no persisten signos radiológicos de hiperostosis.
La formación de grandes cantidades de nuevo hueso subperióstico también se produce en las fracturas, osteomielitis y escorbuto (18). En estas entidades clínicas el hueso se remodela en dos años, similar a lo que ocurre con los episodios hiperostósicos en la enfermedad de Caffey.
Ninguno de los individuos afectados (III-3, IV-8, IV-9 y IV-11) de las tres generaciones presentaron signos clínicos de osteogénesis imperfecta, como problema principal, es decir, carecían de sordera, dentiogénesis imperfecta, escleras azules y ausencia de historia de fracturas (5,17). El diagnóstico es clínico-radiológico, y la radiografía simple del segmento corporal afecto es el método de imágenes indicado.
Conclusiones
Los hallazgos descritos en este artículo permiten reafirmar el fenotipo de las entidades relacionadas con cambios genéticos en los genes de colágeno tipo I, e incluye el trastorno de hiperostosis cortical infantil.
Es necesario conocer los aspectos relevantes de la entidad clínica, a fin de que sea diagnosticada acertadamente. A su vez, se debe considerar que los signos y síntomas de la hiperostosis son sutiles y solo se expresan en la edad preescolar, lo cual hace difícil el diagnóstico en manos inexpertas; por ende, podrían perderse algunos casos aparentemente no afectados en grado severo.
Es imprescindible un adecuado y minucioso examen físico del caso índice, complementado con la interpretación de la genealogía, que es un instrumento que ayuda a orientar el diagnóstico, y el estudio molecular lo corrobora.

