











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkRevisión de tema
La enfermedad de Von Willebrand, causada por una deficiencia del factor de Von Willebrand, es de herencia autosómica, codificada por un gen del cromosoma humano 12 y causada por mutaciones que llevan a un déficit en la síntesis o función de este. Dicha patología es de una alta prevalencia. En dos grandes estudios prospectivos epidemiológicos se encontró en hasta el 1 % de la población general (1). Se ha calculado que la prevalencia de las manifestaciones más graves de la enfermedad (EVW tipo 3) es de entre 1 y 3 por millón (2), y se manifiesta aproximadamente dos veces más en mujeres que en hombres (1).
Descubierta en 1926 por Erik von Willebrand, médico pediatra finlandés, quien centró su interés en una niña de 5 años de edad, cuya familia tenía una larga historia de sangrados. Estos se habían presentado en 23 de las 66 mujeres de esta familia (3,4), una de quienes sangró hasta morir durante la adolescencia, secundario a su periodo menstrual. Entonces esta patología se describió por primera vez como un trastorno hemorrágico hereditario diferente a la hemofilia.
Durante los años cincuenta y principios de los sesenta se demostró que este trastorno estaba relacionado con un nivel reducido de la actividad procoagulante del factor VIII (FVIII) y que mejoraba con la infusión de plasma; pero solo hasta 1971 se demostró que el FVIII y el factor Von Willebrand (FVW) eran proteínas distintas (5,6).
El FVW es una glicoproteína multimérica sintetizada por las células endoteliales y los megacariocitos. Este se guarda en los gránulos alfa de las plaquetas y en las células endoteliales, logrando un papel importante en la hemostasia primaria, ya que ayuda a las plaquetas a aglutinarse y adherirse a la pared de los vasos sanguíneos, para formar así un puente de adhesión entre las plaquetas y el tejido subendotelial vascular y el sitio de daño endotelial, lo cual es necesario para la coagulación normal de la sangre. Además, ayuda al FVIII a prolongar su vida media al unirse a él y así formar el coágulo de fibrina, dado que lo protege de una inactivación proteolítica por parte de las proteínas C y S (7,8,9,10).
Existen varios tipos de esta patología (se clasifica en tres tipos) y su severidad en el sangrado es proporcional al grado de la deficiencia primaria del FVW o secundaria del FVIII (5). La más frecuente (75 %) es la tipo 1 y ocurre por un déficit cuantitativo parcial del FVW y del FVIII. La tipo 2 es por déficits cualitativos (es decir, su función), y se diferencia en cuatro diferentes formas de padecimiento, entre las que se encuentran: tipo 2A (disminución de la función dependiente de las plaquetas, con multímetros anormales), tipo 2B (que se manifiesta con un incremento de la afinidad para la fijación plaquetaria), tipo 2M (en la que se observa una disminución de la función plaquetaria, con multímeros normales) y tipo 2N (en la que la fijación al FVIII se encuentra disminuida), y la tipo 3, que es la menos común y la más severa y ocurre por un déficit cuantitativo severo o ausencia del FVW y del FVIII (11,12).
Las principales manifestaciones de esta patología son sangrados tras cirugías (manifestación usual de las coagulopatías) y epistaxis o metrorragia (manifestación usual de la trombocitopenia) y las hemorragias uterinas anormales por esta patología FVW se encuentra entre el 11 y el 20 % (13). Los criterios diagnósticos se basan en:
Antecedentes familiares de hemorragias excesivas o con el diagnóstico.
Manifestaciones clínicas de los tipos 1 y 2, como epistaxis-gingivorragia-menorragia-hemorragia de las vías digestivas, sangrados abundantes en procedimientos quirúrgicos al que hayan sido enfrentados.
Manifestaciones clínicas en la tipo 3, como hemartrosis o hematomas.
El diagnóstico de certeza se establece con exámenes de laboratorio (14,15) complementarios; con la prolongación del tiempo de tromboplastina parcial (PTT) vía extrínseca de la coagulación; el recuento plaquetario, que usualmente es normal, y con exámenes paraclínicos especiales: actividad del cofactor de ristocetina o actividad fijadora del colágeno (FVW:RCo o FVW:CB), el cual evalúa la habilidad del FVW para aglutinar plaquetas uniéndose al receptor GPIb, en la presencia de ristocetina; los niveles de antígeno de Von Willebrand, la actividad del FVIII y la agregación plaquetaria inducida por ristocetina (RIPA). En conclusión, se mide la actividad funcional del FVW. Los niveles normales del FVIII y el cofactor de ristocetina son mayores a 50 UI/dl, y los niveles diagnósticos son < 30 UI/dl (16,17,18).
El tratamiento de la enfermedad de Von Willebrand se divide en dos tipos de terapias: las coadyuvantes, que proporcionan un beneficio hemostático indirecto (por ejemplo, los antifibrinolíticos, el ácido tranexámico y el ácido épsilon-aminocaproico), y las aplicaciones de preparaciones hemostáticas tópicas, como sellador de fibrina en intervenciones dentales. En las mujeres con menorragia, la administración de una terapia hormonal con anticonceptivos orales combinados es muy útil para regular el ciclo menstrual y controlar el crecimiento endometrial, a fin de producir sangrados más cortos. También se usan sistemas intrauterinos con progesterona (como Mirena®), pues evitan que el endometrio responda al estradiol circulante, independientemente de la función ovárica, y haya una menor pérdida sanguínea menstrual.
En segundo lugar, están los tratamientos que incrementan las concentraciones plasmáticas de los factores de la coagulación de manera aguda (FVW y FVIII) (16), el principal: la desmopresina (30 μg/kg), menos en la enfermedad de Von Willebrand tipo 3, porque es un análogo sintético de la hormona antidiurética, que conserva propiedades antidiuréticas, pero no vasoactivas. Su pico máximo hemostático ocurre a las 0,5 y 1 hora después de su administración. Acorta el tiempo de tromboplastina parcial activada prolongado y el tiempo de sangrado, efectos que promueven la liberación del FVW/FVIII de 3 a 5 veces arriba de las concentraciones basales. La desmopresina no tiene efectos en el recuento o la agregación plaquetaria; pero sí aumenta su adhesión a las paredes vasculares.
Cuando este tratamiento está contraindicado o no es eficaz, la opción terapéutica es la transfusión del FVW y del FVIII. Sus concentraciones se pueden restablecer mediante la infusión de estos concentrados (se utiliza en los déficits severos como en la enfermedad de Von Willebrand tipo 3), manteniendo la actividad del FVIII y de FVW a un 50-100 % (19).
Cambios hematológicos durante embarazo
En un embarazo de evolución normal se evidencian cambios fisiológicos, y dentro de estos hay alteraciones de la hemostasia, causada por un aumento en la concentración de algunos factores de la coagulación como son el FVIII y el FVW, así como la disminución de la actividad fibrinolítica (20). Estos cambios mantienen la función placentaria, pero pueden predisponer a trombosis y complicaciones vasculares de la placenta (21).
Los factores de la coagulación mencionados aumentan a partir del segundo trimestre, hasta llegar a concentraciones > 30,0 UI/dl y se mantienen hasta el término del embarazo (pico máximo). En la enfermedad de Von Willebrand tipo 1 pueden llegar a ubicarse dentro de rangos normales; en la tipo 2 presenta un aumento más impreciso. Estos cambios fisiológicos no ocurren en la enfermedad tipo 3, en la cual los niveles séricos del FVW se depletan rápidamente en el posparto inmediato, potenciando el riesgo de hemorragia postparto. Estas variaciones de la enfermedad nos muestran que la respuesta hemostática en el embarazo depende del tipo y de la severidad de la enfermedad (21). Finalmente, hay un descenso de las hormonas, el estradiol en primer lugar, lo que explicaría la reducción de los niveles del FVW en el posparto inmediato (22,23,24,25).
Los estudios hablan de un 33 % de metrorragia del primer trimestre, y el 10 % se presenta con hemorragias severas que requieren transfusión. Al clasificar esta patología, la enfermedad tipo I tiene una incidencia de hemorragia posparto del 18,5 %, la tipo 2 llega hasta un 20 % (25) y el riesgo es mayor en las de tipo 2 y 3, con un pico máximo entre los 12 y 20 días, persistiendo hasta 5 semanas posparto. Hay un riesgo: en la anestesia regional, en la cual hay una alta posibilidad de producir hematoma espinal que puede hacer compresión medular (26,27).
Las complicaciones hemorrágicas durante el embarazo son más frecuentes cuando los niveles del cofactor de ristocetina de Von Willebrand y los niveles del FVIII son < 50 UI/dl. Por eso, se debe realizar seguimiento de su actividad dos semanas antes del parto y del posparto. En el momento de la atención del parto se debe hospitalizar a la paciente en una institución de tercer o cuarto nivel de complejidad, donde se puedan monitorizar niveles de FVIII y FVW, instaurar la terapia ante cualquier evento invasivo y brindar profilaxis con FVW a todas las pacientes con enfermedad tipo 3. No se usa tanto su administración en la tipo 1 si los niveles FVIII y FVW > 50 UI/dl en el parto o hasta 3 a 5 días posparto.
Si se presentan episodios de sangrado, deben tratarse con FVW o con desmopresina (DDAVP) y continuar el tratamiento con DDAVP o FVW de manera intermitente dos a cuatro semanas después del parto. Los niveles de FVIII se deben monitorizar durante tres días y medio posparto y hacerles seguimiento por dos semanas. Solo en pacientes con FVW (cofactor de ristocetina o FVIII) por debajo del rango terapéutico está indicada la profilaxis con agentes hemostáticos (27,28,29).
Presentación del caso clínico
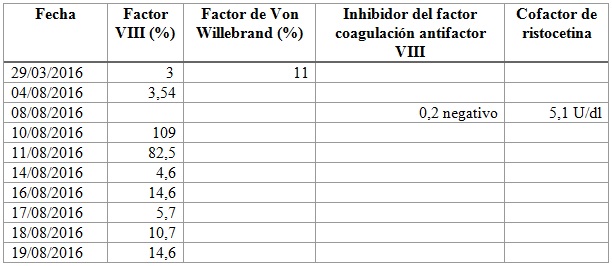
El caso clínico corresponde a una mujer de 18 años de edad con un embarazo de 38,5 semanas de gestación que ingresó el 3 de agosto de 2016 con diagnóstico de enfermedad de Von Willebrand tipo 3, diagnosticada desde los 3 años de edad, manejada de manera profiláctica con FVIII, dos veces a la semana. Ingresó a la institución para inducirle el trabajo de parto; por ello, se le tomaron exámenes paraclínicos previos y se inició el control de la enfermedad de base en manejo conjunto con el servicio de hematología. Se estableció el esquema de manejo durante el trabajo de parto y puerperio de la siguiente manera:
Meta de actividad del factor mayor a 50 U/ml.
Administrar 3000 UI de FVIII plasmático (inmunate), una vez se encuentre en trabajo de parto en la fase activa.
Administrar 1000 UI de FVIII plasmático (inmunate) a las 12 horas del posparto.
Administrar 1000 UI de FVIII plasmático (inmunate) al día por 3 a 5 días del posparto según la evolución.
Medición diaria de la actividad del FVIII para el ajuste de la dosis.
Se inició la inducción del trabajo de parto el 8 de agosto con misoprostol y una adecuada respuesta y evolución del trabajo de parto. El parto eutócico se atendió a las 14:30 horas, del cual se obtuvo un recién nacido vivo, con desgarro perineal grado I y adecuado control del tono y del sangrado uterino. De acuerdo con el servicio de hematología, se manejó con FVIII durante la fase activa y el puerperio, con medición de niveles del FVIII durante 3 a 5 días luego del parto dentro de metas mayor a 50 UI/dl y actividad mayor al 50 %, por lo que se le dio egreso sin profilaxis por indicación de hematología.
Reingresó el 14 de agosto con sangrado abundante, encontrándose anemia y un descenso de 3 g de hemoglobina (7,3 g/dl), taquicardia y episodios de lipotimia. Se trasladó a la sala de partos, donde se activó el código rojo a las 5 a. m., con hemorragia posparto tardía secundaria a coagulopatía (enfermedad hematológica de base) y choque hipovolémico moderado. Se inició manejo de código rojo, según el protocolo, con líquidos endovenosos oxitócica 10 UI/h y transfusión de 3 unidades de glóbulos rojos empacados (UGRE). Se recibieron los exámenes paraclínicos que reportaron prolongación de la PTT, la anemia descrita, plaquetas normales y el fibrinógeno consumido (260 mg/dl). Se administraron 2000 U de FVIII a las 6 a. m. Nuevamente, hacia las 9:30 a. m., presentó sangrado moderado de 300 cm3 con coágulos, por lo que se le administró misoprostol, y hacia las 11 a. m. nuevamente hubo sangrado de 200 cm3. Por ello, se decidió colocarle un balón de Bakri con 300 cm3 de llenado y administración de 0,3 µg/kg de desmopresina; procedimientos realizados sin complicaciones con control de sangrado. Con los exámenes paraclínicos de control realizados 6 horas postransfusión se halló corrección de la PTT, plaquetas normales, con bajo rendimiento transfusional con hemoglobina en 7,7 g/dl con persistencia de taquicardia. Ello indicó la realización una nueva transfusión de 2 UGRE y traslado a la unidad de cuidado intensivo (UCI).
Durante estancia en la UCI (3 días), presentó una adecuada evolución clínica con remisión de la taquicardia y del sangrado. Se le retiró el balón de Bakri 24 h después, con una adecuada respuesta, sin nuevos episodios de sangrado genital. Tuvo un nuevo descenso de los valores de hemoglobina de 1 g (hemoglobina en 6,4 g/dl), por lo que se le transfundió una UGRE más y se trasladó a ginecología. Evolucionó favorablemente con hemoglobina postransfusión en 9,2 g/dl y manejo con 2000 UI/12 h de FVIII, con niveles de este en ascenso. La paciente fue valorada por el servicio de hematología, que consideró que tenía una adecuada respuesta al manejo instaurado y, así, le dieron egreso con profilaxis con FVIII, 2 veces por semana, asegurándose que la empresa promotora de salud se la suministrara en su lugar de procedencia (Chámeza, Casanare).
Discusión
La enfermedad de Von Willebrand es el trastorno hemorrágico hereditario más común. Se encuentra en aproximadamente en el 1 % de la población general, aun cuando muchas de sus presentaciones son subdiagnosticadas, por presentarse en formas leves; por lo que es indispensable una adecuada historia clínica en la cual se consignen antecedentes familiares y personales, así como un buen examen físico, y de sospecharse esta patología, confirmar con laboratorios. Si se trata durante el embarazo, se clasificará como de alto riesgo, y definitivamente se debe manejar en conjunto con el servicio de hematología para disminuir su morbilidad, y a fin de garantizar una adecuada atención periparto, se deberían monitorizar las concentraciones de FVW y FVIII durante el tercer trimestre del embarazo, con el propósito de lograr un parto seguro. Es preciso también educar a las pacientes respecto a la posibilidad de hemorragias posparto, que requieren consulta inmediata.
Los obstetras tenemos que estar atentos a las hemorragias posparto a las que estamos expuestos y estar preparados para el manejo adecuado en caso de que se trate de una coagulopatía hereditaria, como la enfermedad de Von Willebrand, a efectos de minimizar los riesgos y las complicaciones maternas y neonatales, entendiendo que el manejo es claramente multidisciplinario. Los mayores riesgos son sufrir hemorragia posparto primaria (> 500 ml de pérdida de sangre en las primeras 24 h) y secundaria (24 h a 6 semanas después del parto), debido a la rápida caída en las concentraciones de FVIII y FVW después del parto. El riesgo de hemorragia posparto puede reducirse mediante el manejo activo de la tercera etapa del parto y minimizando el trauma genital y perineal materno.
En pacientes con la enfermedad de Von Willebrand, tipos 1 o 2, las cantidades del FVW y del FVIII aumentan de 2 a 3 veces durante el segundo y tercer trimestres; pero descienden a los niveles iniciales poco después del parto. Las mujeres con la enfermedad de Von Willebrand siempre VWD deben ser monitoreadas para detectar el FVW: RCo y FVIII: C, al menos una vez durante el tercer trimestre del embarazo. El riesgo de sangrado es muy bajo cuando FVIII: C y FVW: RCo los niveles están cerca o más altos que 50 U/dL. En mujeres embarazadas con la enfermedad de Von Willebrand tipo 1 con FVIII: C o FVW niveles menores a 30 U/dl, la administración de desmopresina generalmente se realiza después del pinzamiento del cordón umbilical. El mismo enfoque, con menos infusiones, se puede aplicar a las personas con FVW mayores de 30 e inferiores a 50 U/dl (31).
Es viable usar agentes antifibrinolíticos orales durante este periodo, para prevenir la hemorragia posparto tardía. En las mujeres con la enfermedad de Von Willebrand tipo 3, el FWF y el FVIII no aumentan durante el embarazo y, por lo tanto, pueden requerirse concentrados de FVW/FVIII para controlar el sangrado vaginal intermitente y durante el parto o para la cesárea (32). Es importante verificar los niveles del FVW después del parto en las mujeres con la enfermedad de Von Willebrand, manteniendo el nivel del FVW dentro del rango normal con concentrados que contengan desmopresina o FVW, y durante al menos 3 días después del parto, o 5 días si se realiza una cesárea.
No se encuentra evidencia que contraindique la vía vaginal del parto; sin embargo, se debe individualizar cada caso en particular, para la toma de la conducta, ya que la cesárea debe reservarse solo para las indicaciones obstétricas habituales. Es clave realizar el menor el traumatismo posible tanto para la madre como para el feto, así que se deben evitar o partos instrumentalizados (ventosas, espátulas o fórceps).
Recomendamos permitir el parto espontáneo, siempre que sea posible; prestar especial atención cuando se debe inducir el parto, ya que este es probable que se prolongue y se asocie con la necesidad de parto instrumental o cesárea de emergencia, especialmente en mujeres primigrávidas con cuello uterino desfavorable al inicio de la inducción. En estos casos, un equipo multidisciplinario de obstetra y hematólogo junto con la madre deben realizar una evaluación cuidadosa de los riesgos. En algunas circunstancias, la cesárea electiva podría considerarse menos traumática tanto para la madre como para su hijo afectado.
Diseñar un plan de manejo con el anestesiólogo antes del trabajo de parto podría ser de mucha ayuda para evitar complicaciones. El uso del bloqueo regional en pacientes con trastornos hemorrágicos por esta patología es controvertido, debido al riesgo potencial de hemorragia epidural o espinal y hematoma. Para la tipo 1, si el nivel del FVIII está por encima de 50 UI dl por cada litro, el bloqueo regional no está contraindicado. En situaciones en las cuales los niveles de factor no pueden evaluarse debido al trabajo de parto avanzado, el bloqueo es viable siempre que el nivel de factor sea > 50 UI/dL por cada litro en el tercer trimestre; sin embargo, la anestesia epidural generalmente no se recomienda para su uso en la enfermedad de Von Willebrand tipo 2 o 3 (33).
Hay informes anecdóticos de una disminución de 41 a 9 UI/dL por cada litro en el transcurso de una semana (34). Se reporta, además, que el tiempo promedio de presentación de la hemorragia posparto en mujeres con enfermedad de Von Willebrand fue de 15,7 ± 5,2 días (35). Esto implica la necesidad potencial de profilaxis u observación cercana durante varias semanas después del parto. En caso de sangrados prolongados e intermitentes, se pueden usar el ácido tranexámico o las píldoras anticonceptivas orales combinadas. También se recomienda alentar a los pacientes a informar sobre un sangrado excesivo, para reconsultar, y en el servicio de urgencias documentar las concentraciones de hemoglobina.
Recomendaciones de manejo
El embarazo en mujeres con enfermedad de Von Willebrand lo debe manejar un equipo multidisciplinario que incluya un obstetra, un hematólogo y un anestesista.
Los niveles de factor que incluyen FVW: Ag, FW: Ac y FVIII: C deben verificarse en la reserva, 28 y 34 semanas y antes de los procedimientos invasivos. Se debe administrar un tratamiento profiláctico cuando los niveles de los factores son < 50 UI/dl.
Las mujeres con enfermedad de Von Willebrand tipo I, generalmente, no requieren tratamiento profiláctico para el parto, solo si los niveles de FVW son menores de 30 UI/dl. En la enfermedad de Von Willebrand tipo 2 se necesita tratamiento para el parto quirúrgico o si hay traumatismo perineal. Las mujeres con enfermedad de Von Willebrand tipo 3 precisa tratamiento para todos los tipos de parto.
El parto debe planearse en una unidad donde esté disponible el personal necesario para el manejo de esta patología y, además, tener disponibles los recursos para pruebas de laboratorio, así como los tratamientos.
Las concentraciones del factor deben monitorearse después del parto y la profilaxis para mantener la actividad del FVW y los niveles del FVIII > 50 UI/dl por cada litro durante, al menos, 3 o 5 días después de la cesárea. El ácido tranexámico o la píldora anticonceptiva oral combinada deben considerarse para controlar la hemorragia posparto secundaria prolongada o intermitente.
Según la observación de cómo se desarrolló la evolución de la paciente presentada en el presente caso, en el puerperio tardío, nosotros recomendaríamos que las pacientes con enfermedad de Von Willebrand tuvieran un seguimiento intrahospitalario de al menos dos semanas.

