











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La enfermedad de Von Willebrand (EVW) es un trastorno hereditario que cursa con hemorragias de intensidad variable, causado por alteraciones cuantitativas, cualitativas o funcionales del factor de Von Willebrand (FVW) (1). El FVW es esencial para la unión de las plaquetas al subendotelio de vasos lesionados y para el transporte del factor VIII (FVIII) (2).
La EVW es el trastorno hemorrágico hereditario más común, pues afecta aproximadamente al 1 % de la población (3). Se estima que en Estados Unidos, la prevalencia de esta enfermedad es del 1 % y en Colombia, hasta 2018, representaba la tercera enfermedad huérfana más frecuente con una tasa de 1,4 individuos por cada 100.000 habitantes (4). La mayoría de los casos se transmiten como un rasgo autosómico dominante sin predilección por ningún sexo (5).
A continuación, se hará una revisión general de la EVW hereditaria, con información actualizada acerca de su fisiopatología, diagnóstico y tratamiento, con el objetivo de facilitar su abordaje clínico y paraclínico para el médico de atención primaria.
Metodología
Se realizó una revisión narrativa de la literatura. Se incluyeron las siguientes bases de datos electrónicas en las búsquedas bibliográficas: Medline vía PubMed, ScienceDirect, Clinical Key, Scopus, EBSCO y SciELO. Se realizaron búsquedas con uso de términos MeSH (Medical Subject Headings) y DeCS (Descriptores en Ciencias de la Salud) dependiendo del idioma de publicación de los artículos revisados (inglés y español). Los términos utilizados fueron: enfermedad de Von Willebrand, factor de Von Willebrand, trastornos hemorrágicos, desmopresina. Adicionalmente, se revisaron libros de texto y guías de práctica clínica publicadas por sociedades científicas en el mundo y colombianas, como el Fondo Colombiano de la Hemofilia y el Fondo Nacional de Enfermedades de Alto Costo. Dentro de la revisión se tomaron artículos desde 1987 hasta el 2019, con excepción de la sección de tratamiento, en la que se consideraron únicamente artículos publicados desde 2012 hasta 2019.
Historia
La primera descripción la realizó Erik Von Willebrand, en 1926, quien inicialmente consideró un problema de sangrado mucocutáneo severo presente en una familia del mar Báltico, en la cual varios integrantes fallecieron debido a hemorragias graves de múltiples orígenes (6).
Durante los años cincuenta se descubrió una relación con la disminución de la actividad del FVIII, que lograba ser corregida con plasma de pacientes sanos. Durante la década de los sesenta, el noruego Christian Borchgrevink vislumbró los primeros indicios del rol del FVW en la agregación plaquetaria, información confirmada en 1970 por Margaret Howard y Barry Firkin a través de sus estudios con ristocetina, un antibiótico que induce el proceso de agregación en individuos sanos y hemofílicos, pero no en pacientes con EVW(7). Finalmente, hacia 1985 el gen del FVW es aislado y en 1989 se determina su estructura, gracias a lo cual se confirma que la EVW y la hemofilia A son entidades distintas codificadas por genes independientes (6).
Epidemiología
La EVW es la patología hemorrágica hereditaria autosómica más frecuente. Su prevalencia ha sido estimada en un 1 % (8,9). En población caucásica se estima una frecuencia desde 30 casos por millón de habitantes hasta el 1 % de la población (10). Los casos sintomáticos se estiman en 100 por millón de habitantes. Se estiman 620.000 casos sintomáticos en el mundo, de los cuales aproximadamente el 80 % habita en países en vía de desarrollo (11).
Aproximadamente, 125 individuos por millón presentan EVW sintomática en los Estados Unidos. En un estudio realizado en el norte de Italia informaron una prevalencia del 0,9 %, aproximadamente 8,2 casos por cada 1000 habitantes (12).
Hay tres tipos principales de EVW. El tipo 1 se debe a una reducción cuantitativa en la proteína del FVW; el tipo 2, a un FVW disfuncional, y el tipo 3, a FVW ausente o muy reducido. De los pacientes con EVW, el 70-80 % son tipo 1, el 5-15 % son tipo 2 y menos del 5% son tipo 3. La prevalencia del tipo 3 es de 1 a 5 por millón de habitantes en Europa, y de 3 por millón en Suecia e Israel (13). En Chile y Venezuela representan el 2,71, 1,75 por millón, respectivamente (14).
En Colombia, para el 2018 se reportaron 4271 personas con diagnóstico de coagulopatía, de las cuales el 35,2 % tenía EVW (15,16). En los pacientes con EVW predomina la afectación en el sexo femenino (74,7 %), con una proporción de casi 2:1, probablemente por el sangrado menstrual excesivo, que facilita su diagnóstico (17). La prevalencia en nuestra población es de 3 por cada 100.000 habitantes, con una media de edad de 28 años (DE = 16,4) (16). Es importante tener en cuenta que no se han encontrado preferencias por raza o zona geográfica en el comportamiento de esta enfermedad (9).
Estructura molecular del factor de Von Willebrand
El gen para el FVW se localiza en el brazo corto del cromosoma 12, en la región 13.3. Contiene 178 kilobases (Kb) y 52 exones. Existe, además, un seudogen con copia de los exones 23 al 34 en el cromosoma 22. Codifica para ARNm de 8,8 Kb de longitud, su proteína constituye una glucoproteína plasmática de entre 500 y 2000 KDa, posee 2813 aminoácidos, de los cuales 22 equivalen a un péptido señal, 741 a un propolipéptido (dominios D1 y D2) y 2050 a la subunidad del FVW con propiedades adhesivas necesarias para cumplir su función hemostática (18,19). Se han descrito 140 polimorfismos de inserción y deleción ubicados en su región promotora, regiones intrónicas y exones (6,20,21). Se han observado diferentes tipos de mutación para esta patología, como lo son errores transcripcionales o en la remoción de intrones, mutaciones nonsense, missense, deleciones e inserciones (22).
El FVW es sintetizado por células endoteliales y megacariocitos (20,23), almacenado en gránulos alfa y cuerpos de Weibel-Palade (23). Además, es modulado y clivado por las metaloproteinasas ADAMTS 13 al momento de entrar al torrente sanguíneo (14,20,24). La estructura de esta proteína comprende dominios de la A a la D, estos últimos involucrados en la regulación de la formación multimérica y su adhesión al FVIII de la coagulación. Los dominios A poseen propiedades de fijación al colágeno, y con los dominios C se encargan de la adhesión plaquetaria.
Fisiopatología
La EVW se caracteriza por defectos cuantitativos o cualitativos del FVW (25). Este último desempeña una papel fundamental en la localización e inicio de la respuesta hemostática primaria y secundaria (26,27).
Posterior a una lesión endotelial, hay incremento en la concentración local del FVW, por su unión a componentes de la matriz subendotelial, como el colágeno. Esta interacción facilita la unión del FVW al receptor del complejo Ib-IX-V de glicoproteína plaquetaria (GPIb-IX-V); adicionalmente, el estrés por cizallamiento produce un cambio en la forma del FVW, facilitando su unión a la glicoproteína Ib plaquetaria (GPIb) (28). Ello permite la adherencia de las plaquetas circulantes al sitio lesionado, al promover su agregación y favorecer la formación del tapón plaquetario (figura 1) (29). La ausencia del FVW o una estructura anormal dificulta el proceso de reclutamiento plaquetario y determina una hemostasia primaria subóptima (30,31).
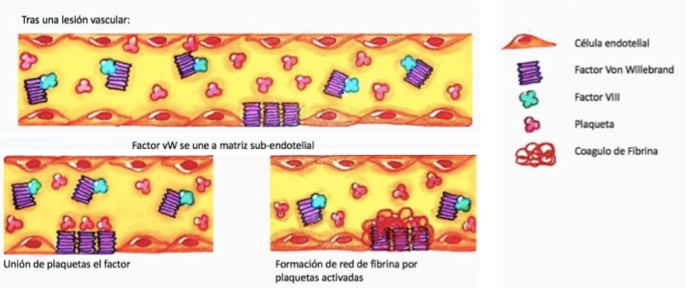
Por otro lado, el FVW es responsable de la protección y transporte del FVIII, pues asegura su contacto con los fosfolípidos de superficie de plaquetas activadas y endotelio lesionado (32). En este sentido, la ausencia del FVW implica una disminución en la vida media del FVIII, lo que genera alteraciones en la hemostasia secundaria (33).
El FVW es el transportador plasmático del factor VIII. Luego de una lesión endotelial, el FVW se une a la matriz subendotelial, y gracias a fuerzas de cizallamiento, sufre un cambio conformacional que permite la adherencia plaquetaria mediante el receptor glicoproteico GPIb. Las plaquetas son activadas e inician el proceso de agregación, alterando sus fosfolípidos de superficie y facilitando la unión del factor VIII. Este último en su interacción con el factor Xa favorece la cascada de coagulación y formación final del coágulo de fibrina.
Clasificación
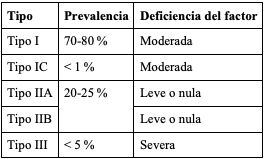
La EVW se clasifica en diferentes tipos (tabla 1), que se explican a continuación (34,35).
Tabla 1. Prevalencia y nivel de deficiencia del factor de Von Willebrand en los tipos de enfermedad de Von Willebrand
Alteraciones cuantitativas
Tipo 1. Es la forma más frecuente de la enfermedad. Se caracteriza por una disminución en la cantidad del FVW en la sangre, así como una menor actividad plaquetaria dependiente de este (23). Es heredado de manera autosómica dominante, con mutaciones que afectan la producción en las etapas de empaquetamiento y secreción, lo que lleva a una disminución del FVW sérico útil (21).
Tipo 1C. Es una forma extremadamente rara de la enfermedad, en la cual hay una producción adecuada del factor; sin embargo, se metaboliza de manera ultrarrápida, lo que lleva a una disminución en las concentraciones séricas y a una vida media corta de la proteína (23).
Tipo 3. En esta forma de la enfermedad hay una depleción casi completa o completa del factor, acompañada de deficiencia de FVIII (36). Esto genera formas muy severas de la enfermedad, como sangrados articulares o musculares profundos. Es la forma menos prevalente (37), y la mayoría de pacientes con este tipo de la enfermedad muestran mutaciones recesivas o heterocigotas compuestas (23).
Alteraciones cualitativas
Tipo 2A. Esta forma la causa una deficiencia en la formación de FVW funcional, que se da por una disminución o ausencia de multímeros de alto peso molecular o por la formación de multímeros anormales previo a la liberación de la proteína (31). Si bien el problema no se encuentra en la cantidad de FVW, este se puede encontrar disminuido (38).
Tipo 2B. Se caracteriza por una mutación que genera mayor unión entre el dominio AI del FVW y la GPIb-alfa (39). In vivo la unión exagerada de estas dos moléculas aumenta su barrido y lleva a una disminución en la cantidad de FVW. En esta forma de la enfermedad, algunos pacientes cursan con trombocitopenia, lo que se consolida como un factor de riesgo adicional para sangrado (40).
Tipo 2M. Este subtipo de la enfermedad se caracteriza por una mutación de la herencia autosómica dominante en la cual el FVW posee todos su multímeros; pero tiene una baja afinidad, lo que impide la unión plaquetaria (34,35). Clínicamente, se ha visto que este tipo puede ser equivalente a la tipo IIB (41).
Tipo 2N. Este subtipo está dado por una mutación autosómica recesiva que puede darse en los exones 18-20 o en el sitio de clivaje por la furina para la remoción del propéptido (8) en el sitio de unión al FVIII, por lo que genera una disminución de la afinidad del FVW al factor VIII y, por consiguiente, disminución de sus concentraciones plasmáticas. Este mecanismo fisiopatológico explica por qué este tipo puede simular el cuadro clínico de la hemofilia tipo A (21,41,42).
Seudo-Von Willebrand
El término seudo-Von Willebrand se refiere a una patología dada por una mutación autosómica dominante en la que hay una mutación missense en el codón 239, que remplaza una metionina por una valina y que lleva a la formación de un receptor GPP IB hiperfuncionante (43). El cambio estructural del receptor lleva a que este se encuentre constantemente activado y, por lo tanto, se mantenga unido al FVW, por lo que simula la EVW con un FVW sin ninguna alteración (1).
Cuadro clínico
La EVW se caracteriza por cursar con sangrados frecuentes; sin embargo, usualmente médicos y pacientes desatienden este síntoma y así se pueden tardar años en obtener el diagnóstico correcto; por esto, se debe tener una alta sospecha clínica de la enfermedad si se presentan sangrados a repetición, especialmente mucocutáneos como: epistaxis, de la mucosa oral, púrpuras, petequias, sangrados gastrointestinales, y en las mujeres jóvenes en edad reproductiva, sangrados menstruales abundantes (23).
La presentación del cuadro clínico depende del tipo de enfermedad del paciente. Por lo general, las formas más severas se diagnostican en la infancia o adolescencia; mientras que las formas leves se diagnostican en pacientes adultos (44). Los signos y síntomas de la EVW más frecuentes en mujeres en edad reproductiva son (45):
La presencia de coágulos en el flujo menstrual de más de 2,5 cm de diámetro.
La necesidad de cambiar la toalla higiénica o el tampón cada hora o con mayor frecuencia.
Síntomas de anemia: cansancio, fatiga o disnea.
A pesar de ser una patología que presenta sangrados como síntoma principal, es necesario diferenciar entre aquellos dados por trastornos de la coagulación y otras patologías (46). Se han diseñado cuestionarios para dar una puntuación según los síntomas, denominados bleeding score (47), como herramienta para cuantificar las manifestaciones y contribuir a discriminar entre sujetos con EVW y sin esta, y un cuestionario y score pediátricos. Para definir sangrado menstrual excesivo, se utiliza el Pictorial Bleeding Assessment Chart (48).
Diagnóstico
Hacer un correcto diagnóstico de la EVW es fundamental, por lo que es necesario basar la evaluación clínica en tres pilares: la historia personal, la historia familiar y la evaluación paraclínica.
Historia personal
Es necesario indagar específicamente por síntomas hemorrágicos como sangrados mucocutáneas excesivos, hematomas sin motivo aparente, epistaxis y laceraciones con hemorragias excesivas o de difícil control (41). En las mujeres es fundamental hacer una detallada descripción de los patrones menstruales, ya que esta puede ser la única manifestación de la enfermedad (49).
Historia familiar
Se deben evaluar los antecedentes familiares de hemorragias excesivas. Si bien es común encontrar antecedentes familiares, dejar de hallarlos no descarta la patología, ya que existen algunas presentaciones de la enfermedad que tienen un patrón hereditario autosómico recesivo y grados de penetrancia variable (50).
Exámenes paraclínicos
El tamizaje de la EVW es limitado, por la baja fiabilidad de los exámenes utilizados. El tiempo de tromboplastina parcial puede encontrarse normal en casos de EVW de tipo 1 leve o prolongado en caso de niveles bajos de FVIII, lo que restringe su uso como herramienta de cribado (51). Por otro lado, a pesar de su baja sensibilidad y especificidad, los análisis de función plaquetaria deben contemplarse en el abordaje diagnóstico de la enfermedad (38,52,53).
Considerando que no se cuenta con una prueba sencilla y aislada que permita diagnosticar la EVW, debe utilizarse una batería de pruebas de laboratorio (tabla 2), cuyos hallazgos varían dependiendo del tipo de EVW que padece el individuo (tabla 3). Adicionalmente, se encuentran disponibles pruebas de laboratorio específicas que contribuyen a confirmar el tipo y subtipo de EVW (tabla 4). El gen del FVW tiene múltiples variaciones de la secuencia benigna y sus deleciones amplias no se detectan en la secuenciación habitual de ADN, lo que dificulta el diagnóstico genético (54).
Tabla 2. Pruebas de laboratorio en el diagnóstico de la enfermedad de Von Willebrand
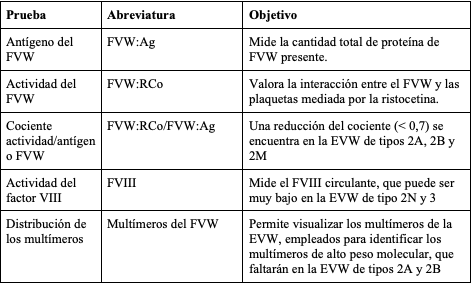
adaptado de (52).
Tabla 3. Hallazgos de laboratorio esperados en cada tipo de la enfermedad de Von Willebrand
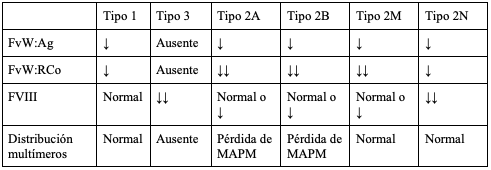
FvW:Agantígeno del FVW
FvW:RCoactividad del cofactor de ristocetina del FVW
FVIIIfactor VIII
MAPMmultímeros de alto peso molecular
adaptado de (52)
Tabla 4. Pruebas de laboratorio especializadas en la enfermedad de Von Willebrand
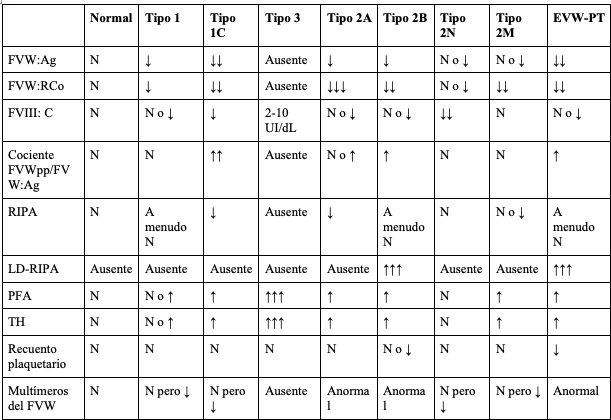
FVW:Agantígeno del FVW
FVW:RCoactividad del FVW por el cofactor de ristocetina
FVIII:Cactividad coagulante del factor VIII
FVWpppropéptido del FVW
RIPAagregación plaquetaria inducida por ristocetina
LD-RIPAagregación plaquetaria inducida con dosis bajas de ristocetina
THtiempo de hemorragia
EVW-TPenfermedad de Von Willebrand de tipo plaquetario
Nnormal
N pero ↓normal pero de intensidad reducida
adaptado de (52)
Durante la valoración paraclínica inicial del paciente con sospecha de EVW se tiene como objetivo detectar la presencia o ausencia del FVW (FVW:Ag), sus productos en la cascada de coagulación (FVIII:C) y su capacidad de unión o no a GPIba mediado por ristocetina (FVW:RCo). Por otro lado, las pruebas de segunda línea incluyen el análisis del patrón multimérico de la proteína, que brinda información acerca de su estructura como es el caso de la EVW tipo 2A y 2B (tabla 2) y el análisis de RIPA (prueba de agregación plaquetaria inducida por ristocetina entre 0,7 y 1,2 mg/ml), que detecta alteraciones en la respuesta a la ristocetina, como en la EVW 2A y 2M (55).
Se encuentran disponibles otras pruebas especializadas como LD-RIPA (< 0,7 mg/ml), que evalúa la respuesta de agregación plaquetaria a bajas dosis de ristocetina, provocando su aglutinación como en el caso de la EVW tipo 2B y el cociente entre la determinación inmune del propéptido (FVWpp) y FVW:Ag (FVWpp/FVW:Ag), que constituye un marcador de depuración acelerada del FVW (56). Cuando se evidencia un aumento > 2-3 en dicho cociente, es utilizado para distinguir una manifestación severa de EVW tipo 1 de la EVW tipo 3 (53). En la figura 2 se resume en un algoritmo las indicaciones para el diagnóstico de la EVW y sus subtipos (57,58).
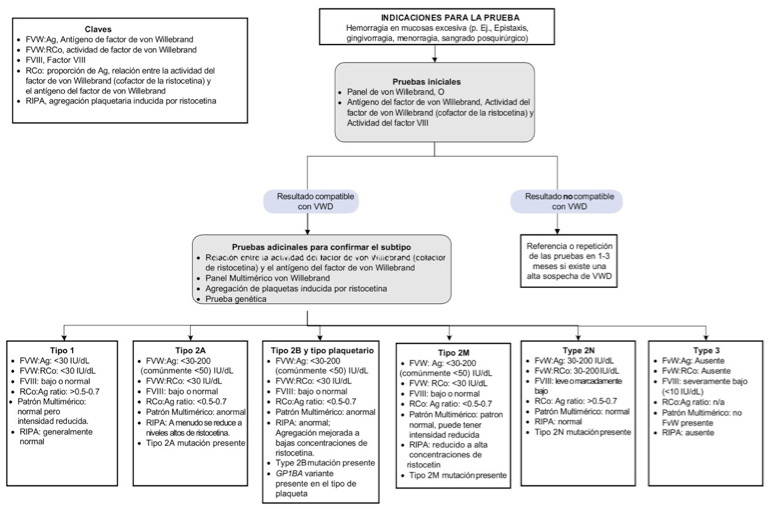
adaptado de (57)
Figura 2. Algoritmo diagnóstico para la enfermedad de Von Willebrand y sus subtipos
Las pruebas iniciales en pacientes con sospecha de trastornos hemorrágicos suelen incluir el hemograma, recuento de plaquetas, tiempo de protrombina (PT) y tiempo parcial de tromboplastina activada (aPTT). Sin embargo, tienen un uso limitado en el diagnóstico de EVW porque son normales en la mayoría de los subtipos, no son sensibles, ni específicas para la EVW. Si el paciente no tiene anomalías detectadas por las pruebas de hemostasia iniciales se recomienda utilizar el algoritmo indicado.
Tratamiento
En pacientes con EVW, el objetivo terapéutico es corregir el defecto dual de la hemostasia. Las opciones terapéuticas incluyen el uso de desmopresina, que libera FVW endógeno de las células endoteliales y el FVW exógeno de los concentrados derivados de plasma de FVW/FVIII. Esto logra incrementar los niveles de FVW, por lo que previenen eventos hemorrágicos severos (59).
Desmopresina (1-ácido8-D-arginina vasopresina) (DDAVP). Es un derivado sintético de la hormona antidiurética que aumenta las concentraciones plasmáticas de FVW y FVIII. Se cree que este efecto se da por la activación del endotelio vascular, que libera el FVW desde sus zonas de almacenamiento y mejora los niveles de FVIII al evitar su degradación (60). El FVW y FVIII aumentan de tres a cinco veces por encima de los niveles basales cuando se inyecta DDAVP por vía intravenosa (IV), subcutánea (SC) o intranasal (61). La dosis IV es de 0,3-0,4 µg/kg, máximo 20 µg. Se infunde diluida en 50 a 100 ml de solución de NaCl al 0,9 % en 20 a 30 min. Por vía intranasal la dosis recomendada es de 150 a 300 µg (62).
Los pacientes con EVW tipo 1 presentan mejor respuesta al uso de DDAVP. En la EVW tipo 2 la respuesta se correlaciona con el tipo de defecto congénito: para el subtipo 2A se necesita la prueba terapéutica de DDAVP, porque habrá liberación de FVW con multímeros de alto peso molecular disfuncionales. En la variante 2B se encuentra contraindicada, por trombocitopenia secundaria a la agregación plaquetaria. En la 2M se espera una respuesta escasa por alteración en el receptor de unión a la plaqueta. En el subtipo 2N ocurre un incremento transitorio del FVIII, al no poder ser estabilizado por el FVW, por encontrarse mutado el sitio de unión. En la EVW tipo 3 no hay producción endógena de factores, por lo que el uso de DDAVP no genera efecto terapéutico (37).
Debe utilizarse cuando los pacientes van a someterse a procedimientos con riesgo de sangrado leve a moderado. Adicionalmente, se encuentra indicado en pacientes con sangrado menstrual excesivo o epistaxis. Deben tenerse en cuenta los posibles efectos adversos como la cefalea, el enrojecimiento facial y la hiponatremia, que debe prevenirse evitando la ingesta de líquidos posterior a la aplicación. No se contempla su uso en menores de dos años (23).
Concentrados de FVW/FVIII. Los concentrados derivados del plasma están indicados en pacientes que no responden a la desmopresina, que requieren hemostasia mayor o mantener el efecto hemostático por al menos 2-3 días. Los productos que contienen FVW inicialmente se prescribieron para el tratamiento de la hemofilia, y la cantidad de FVW dependía, en gran medida, del proceso de purificación de cada producto (63). Existen concentrados puros de FVW que aumentan inmediatamente los niveles de esta molécula sin incremento inmediato del FVIII; se prefieren estos para procedimientos programados. Para los sangrados severos o intervenciones de urgencia es necesario aplicar concentrados duales que contienen ambos factores. Es necesario familiarizarse con los diferentes coeficientes de los productos disponibles y basar la elección en los niveles basales del paciente (23). La elección del compuesto se basa en su cantidad de FVW y de FVIII. En situaciones de hemorragia severa se prefieren agentes con mayor cantidad de FVIII seguidos de mantenimiento con compuestos con alta cantidad de FVW (preparados recombinantes de FVW) (63).
Hay varios productos disponibles que contienen FVW en alta concentración (64):
La dosis y la frecuencia de administración se deciden teniendo en cuenta el peso del paciente, el tipo y la gravedad de los sangrados o intervención quirúrgica y la función de control de los parámetros clínicos y analíticos pertinentes (65). En general, el objetivo es mantener la actividad del FVIII y el FVW (medido como actividad del cofactor de ristocetina [FVW:RCo]) entre el 50 y el 100 % para hemorragias graves o cirugía mayor. La infusión de 20-50 unidades internacionales (UI)/kg de actividad del cofactor de ristocetina eleva la concentración plasmática a 50 y a 100 %, o 0,7 unidades/ml (70 %); se administra cada 12 a 24 horas. Una dosis de carga de 50 a 80 UI/kg se administra al comienzo del tratamiento para asegurar que se alcance el nivel deseado (66).
Adicionalmente, deben tenerse en cuenta las recomendaciones para procedimientos quirúrgicos: cirugía menor, niveles de FVW:RCo ≥ 50 UI/dL el día del procedimiento y el primer día postoperatorio, y > 30 UI/dL 2 a 5 días postoperatorio, o hasta la caída de la escara. Cirugía mayor, los niveles de FVW:RCo deben ser 100 UI/dL el día del procedimiento y durante el primer día postoperatorio, y deben mantenerse ≥ 50 UI/dL de 7 a 14 días o hasta la cicatrización completa (67).
Profilaxis
Actualmente, no existe consenso de las indicaciones para dar profilaxis a los pacientes con EVW. Se han desarrollado múltiples estudios en su mayoría en el marco de The Von Willebrand Disease Prophylaxis Network (68). Los estudios realizados han demostrado que la administración de concentrado de FVW-ristocetina en dosis que varían entre 39 a 60 U FVW:Rco/kg a largo plazo logra reducir eventos hemorrágicos (epistaxis, hemartrosis, sangrados gastrointestinales) y mejora la calidad de vida, disminuyendo el tiempo de estancia hospitalaria y el ausentismo escolar (69).
Terapias coadyuvantes
Algunas situaciones ameritan adicionar agentes antifibrinolíticos como el ácido tranexámico o el ácido épsilon aminocaproico. Están indicados como terapia coadyuvante en situaciones de hemorragia leve-moderada, procedimientos dentales, sangrado menstrual excesivo y epistaxis (70).
Ambos medicamentos son ácidos gamma-amino-carboxílicos análogos de la lisina; actúan inhibiendo la conversión de plasminógeno a plasmina, que actúa degradando la fibrina, por lo que previene la formación de la red de fibrina y la estabilización del coágulo (71). El ácido epsilón aminocaproico se usa a una dosis inicial de 100 a 150 mg/kg, seguido de una infusión de 10 a 15 mg/kg/h, el ácido tranexámico es 7 a 10 veces más potente, por lo que puede usarse a bajas dosis; dosis inicial de 10 mg/kg seguido de 1 mg/kg/hora (46).
En mujeres con sangrado menstrual excesivo, la administración de terapia hormonal combinada funciona parcialmente, al elevar las concentraciones de FVW y FVIII; el uso de sistemas intrauterinos con progesterona, frecuentemente, aportan beneficios clínicos considerables (1,72).
Discusión
La EVW es el trastorno hematológico más comúnmente heredado, con un patrón de herencia autosómica que afecta al 1 % de la población total. En su mayoría, cursa asintomático y existen variantes graves que pueden comprometer la vida. Para el 2018, en Colombia se documentaron 1504 pacientes con esta patología, con una prevalencia de 3 casos por 100.000 habitantes, clasificándose como una enfermedad huérfana según los lineamientos de nuestro país (16).
Las frecuentes discrepancias entre los resultados de laboratorio, función in vivo y cuadro clínico plantean un reto diagnóstico para el médico, quien debe sospechar esta condición en pacientes con hemorragias frecuentes.
En los últimos años, la caracterización de la estructura del FVW y su producción molecular ha influenciado el diagnóstico paraclínico y el abordaje diagnóstico (73). El entendimiento de los mecanismos biológicos subyacentes aporta a la creación de terapias que mejoran los desenlaces y la calidad de vida de los pacientes.
Asimismo, la optimización del tratamiento a largo plazo de estos pacientes demanda la conducción de ensayos clínicos, con el objetivo de ofrecer mejor evidencia que permita tomar decisiones clínicas acertadas. Por otro lado, los avances en ingeniería genética resultan prometedores para proveer una terapia genética dirigida a pacientes con EVW (67).
Mientras avanzan las investigaciones es necesario promover la implementación de estrategias de salud pública que mejoren el acceso a la salud ligado a buenas prácticas clínicas, basadas en el trabajo coordinado de un equipo multidisciplinario con los conocimientos y las bases necesarias para brindar la mejor atención posible a nuestros pacientes. Si bien en Colombia existe un lineamiento que precisa los contenidos mínimos para la conformación de programas para la atención integral de pacientes con hemofilia y otros defectos de la coagulación (74), es frecuente el subdiagnóstico de estas condiciones, persiste una brecha importante por disminuir y solo un porcentaje de los pacientes recibe tratamiento en nuestro país. Es prioritario revisar los resultados de las estrategias y acciones movilizadas por los gobiernos de otras naciones de América Latina y el mundo, con el objetivo de implementar las mejores estrategias en nuestro país.
Es fundamental evitar la fragmentación de la atención por medio de los programas de atención integral y supervisión permanente en los pacientes con EVW en aras de reducir la carga de enfermedad producida por esta condición, evitar los desenlaces adversos en salud, mejorar la calidad de vida de los pacientes y optimizar el uso de recursos en el sistema de salud colombiano (74).
La presente revisión documentó la información más relevante y actualizada encontrada en la literatura con el objetivo de llenar los vacíos encontrados en la práctica clínica diaria en nuestro país.

