











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
Las cardiopatías congénitas (CC) son alteraciones de la forma y la función del corazón. Constituyen la segunda anomalía congénita más frecuente, con una prevalencia de 80 casos por cada 10.000 nacidos vivos (1, 2). La frecuencia calculada en Colombia, según Baltaxe y Zarante, es de 1,2 por cada 1000 (3).
La prevalencia aumenta con la edad materna, las enfermedades maternas perinatales, el uso de medicamentos (antibióticos y otros), estatus socioeconómico bajo o ingesta de alcohol, con un OR que varía entre 1,6 y 3,9 (4). Sin embargo solo el 20 % puede atribuirse a síndromes genéticos, exposición a teratógenos y diabetes gestacional, por lo que el 80 % restante corresponde a un origen incierto (2).
En el mundo, las cardiopatías congénitas más frecuentes son la comunicación interventricular (CIV), con 26,2 por cada 10.000; la comunicación interauricular (CIA), con 16,4 por cada 10.000, y el conducto arterial persistente (CAP), con 8,7 por cada 10.000 (1).
En general, entre 1970 y 2017, la prevalencia de las cardiopatías congénitas aumentó, aproximadamente, un 10 % cada 5 años. Debido al desarrollo de la ecografía cardiaca en la década de 1980 (5), los estudios perinatales y su reporte internacional han permitido atribuir hasta un 90 % de este incremento en el diagnóstico de lesiones leves, usualmente asintomáticas (CIV, CIA y PAD) (2).
En conjunto, estos nuevos datos ubicaron en prevalencia a Asia (82 por 10.000) (6) por encima de Europa (71,8 por 10.000) y Estados Unidos (81,4 por 10.000), siendo las últimas en tiempos pasados las de mayor número de casos (1, 2). En Latinoamérica, la frecuencia de dichas cardiopatías es de 2,6 por cada 10.000 casos, según el reporte del Estudio Colaborativo Latinoamericano de Malformaciones Congénitas, con prevalencias muy heterogéneas entre los países entre 5,9 y 57,4 por cada 10.000 (1). En Colombia se registró una prevalencia de 15,73 por cada 10.000 recién nacidos entre el 2001 y el 2008, la mayoría de ellos (53 %) masculinos (1), con una mortalidad prenatal del 2,7 % y una posnatal del 2,5 % durante el primer año de vida. Los casos con desenlaces fatales sobrepasaron a los reportados en países como Canadá (1,6 % en el primer año de vida).
La mayoría de las cardiopatías congénitas reportadas en Colombia se agrupan alrededor de cinco morfologías principales: CIV (16,5 %), CIA (11,5 %), hipoplasia del ventrículo izquierdo (10,2 %), coartación aórtica (4,4 %) y trasposición de grandes vasos (4,2 %). De estas cardiopatías reportadas, el 64,5 % corresponde a una anomalía de tipo aislado; el 23,5 %, a una de tipo asociado, y el 12 %, a una de tipo complejo (1).
Las cardiopatías congénitas tienen un alto impacto en la morbimortalidad perinatal; el objetivo de este artículo es presentar un caso poco frecuente por la asociación de defectos y su carácter letal.
Presentación del caso
El caso corresponde a una niña recién nacida producto del primer embarazo de una mujer de 37 años de edad, de padres sin consanguinidad, cuya gestación fue de 39 semanas, supervisada, con examen STORCH negativo, ecografía de detalle con arteria umbilical única. En el hospital donde se consultó se le diagnosticó estado fetal insatisfactorio, por lo que indicaron parto por cesárea de urgencia. Recibieron a una recién nacida de 2770 g, en malas condiciones generales con pobre esfuerzo respiratorio, bradicardia e hipotonía; presentó apnea, por lo cual requirió maniobras de reanimación avanzada. Ante el diagnóstico de hipoxia neonatal, la remitieron a nuestra institución para inicio de protocolo de hipotermia terapéutica. La paciente fue recibida en nuestro hospital a las seis horas de su nacimiento.
Se realizaron los siguientes estudios paraclínicos durante la estancia hospitalaria:
Sistema nervioso central: ecografía cerebral: hemorragia de la matriz germinal grado I derecha, con posterior resonancia magnética en límites normales. Cinedeglución con alteración de la fase faríngea y broncoaspiración.
Sistema cardiorrespiratorio: radiografía de tórax con cardiomediastino desviado a la derecha, ecocardiograma que reporta situs solitus en dextroposición, CIA, CIV, conducto arteriovenoso permeable, hipoplasia de la rama pulmonar derecha, persistencia de vena cava superior izquierda e hipertensión pulmonar moderada. Con impresión diagnóstica de síndrome de la cimitarra. Posteriormente, el angio-TAC de tórax fue concordante con estos hallazgos.
En la valoración por genética médica se evidenció una desviación radial de los miembros superiores, en asociación con hipoplasia de los músculos de la masticación por hallazgo incidental en estudio de resonancia magnética cerebral. Como estudio de abordaje inicial, especialmente por la cardiopatía en posible asociación sindrómica, se decidió estudio inicial con Cariofish para cromosomas 13, 18, 21 y X, el cual fue negativo. Una vez descartadas estas cromosomopatías y asociada con cardiopatía con alteración de primer arco faríngeo, se solicitó estudio de hibridación genómica comparativa con microarreglos para descartar síndromes de microdeleciones y duplicaciones, especialmente con sospecha clínica de síndrome por deleción de 22q11.2.
En el quinto día de su estancia, la recién nacida desarrolló síndrome febril, por sepsis neonatal tardía; hubo deterioro progresivo hasta su fallecimiento. Los médicos tratantes solicitaron una autopsia clínica.
En la autopsia se observaron malformaciones menores, dadas por pestañas anormalmente largas, puente nasal ancho, frenillo oral, desviación radial y pliegue palmar único izquierdo. Al realizar la apertura de las cavidades, en el tórax destacó el posicionamiento anómalo del corazón (mesocardio), asociado con un único lóbulo pulmonar derecho, el cual estaba provisto de bronquio fuente.
En el corazón se evidenciaron cuatro cavidades inadecuadamente conformadas, entre ellas, la aurícula derecha, de morfología izquierda con auriculilla foleada, con base de implantación angosta. En su examen interno se apreció un orificio normal para la edad, ingreso de las dos venas cavas e ingreso de un vaso accesorio proveniente del único lóbulo pulmonar derecho, que constituía retorno venoso anómalo (figura 1).
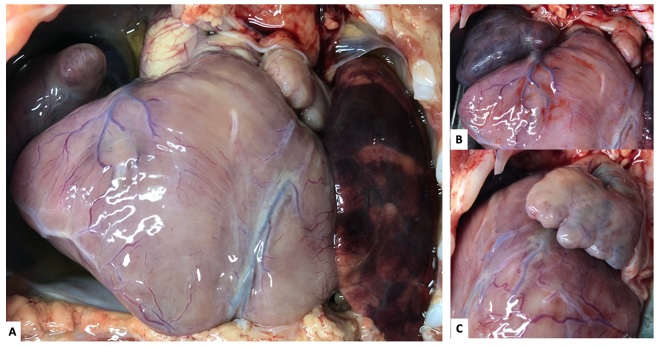
Figura 1 A) Mesocardio. B) Aurícula derecha de base angosta, de morfología izquierda. C) Aurícula izquierda grande triangular, foleada, de morfología derecha.
El ventrículo derecho se encontró inesperadamente liso en su pared septal y no trabeculado. Por el contrario, la aurícula izquierda se observó de gran tamaño, de morfología derecha, y la auriculilla mostró base de implantación amplia. En su examen interior de la aurícula izquierda se apreciaron músculos pectíneos prominentes y abundantes; había drenaje esperado de las venas pulmonares izquierdas. El ventrículo izquierdo mostró pared tortuosa, con músculos papilares prominentes que forman entramados como redes, incluyendo su pared septal. Se evidenció comunicación interventricular tipo membranosa de 0,4 cm de diámetro y la válvula mitral fue de morfología e implantación usual (figura 2).
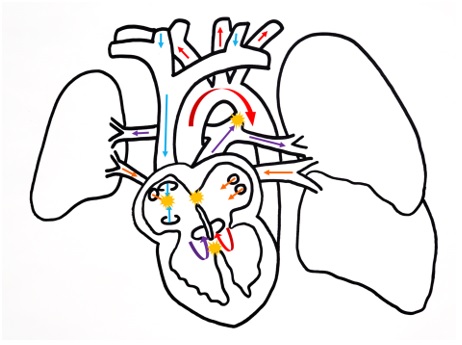
Figura 2. Diagrama de anomalías cardiorrespiratorias. Rojo: flujo sistémico arterial oxigenado; azul: flujo sistémico venoso desoxigenado; naranja: flujo pulmonar oxigenado; violeta: flujo pulmonar desoxigenado; amarillo: cortocircuitos arteriovenosos.
Los sistemas valvulares se apreciaron normales, al igual que los grandes vasos, que tenían orígenes y trayectos usuales.
En el examen de la cavidad abdominal se evidenciaron otras anomalías, dadas por poliesplenia, con bazo accesorio de 0,1 g. Microscópicamente, ambos bazos mostraron hemorragia intersticial. El hígado mostró dos lóbulos accesorios sésiles bilaterales en la convexidad, además de lóbulos derecho e izquierdo simétricos. El páncreas era dismórfico por asimetría. En la cavidad pélvica se observó útero atrésico y el anexo derecho con hipoplasia del ovario y agenesia de trompa uterina (figura 3).
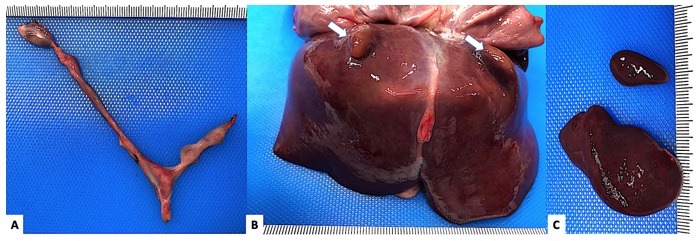
Discusión
El retorno venoso pulmonar anómalo (RVPA) fue descrito por primera vez por Winslow, en 1739 (7). Su característica básica radica en una conexión anormal entre las venas pulmonares y las venas sistémicas, la aurícula derecha o ambas (8). El RVPA es una cardiopatía congénita rara que representa entre el 0,5 % y el 3 % de todas las cardiopatías congénitas (1, 9, 10). Además de su presentación poco frecuente, este grupo de anomalías en el drenaje venoso son poco diagnosticadas prenatalmente, con solo el 1,9 % por ecografía convencional (8), tal y como ocurrió en el caso expuesto.
La etiología de esta anomalía es aún motivo de estudios. Tales anomalías se explican en fallos en la embriogénesis. La aurícula izquierda y las venas pulmonares se desarrollan por separado; por cuanto los brotes pulmonares drenan a un lecho vascular hacia las venas cardinales y umbilicales. La aurícula primitiva forma una vena pulmonar común que luego se une con el lecho vascular pulmonar formado por separado (7). El RVPA se caracteriza por la falla de fusión de una parte o todo del sistema venoso pulmonar con la aurícula izquierda (11); las venas anómalas del lado derecho pueden regresar a la aurícula derecha (como en el caso presentado); la vena cava superior, la vena cava inferior, la vena ácigos, la vena hepática o la vena porta y las venas anómalas del lado izquierdo podrían drenar en la vena innominada, el seno coronario y la vena hemiácigos (12), y los drenajes aberrantes son predominantemente derechos, alrededor del 80 % (7).
Esta CC se subdivide en dos grupos: el primero, una variante completa o total (RVPAT), en la que todas las venas pulmonares tiene un drenaje anormal, de los cuales el 50 % corresponde a formas supracardiacas; el 20 %, a formas cardiacas, y el 30 % restante, a formas infracardiacas (13). El RVPAT más frecuente es el drenaje de la vena cava superior izquierda persistente (14), y de allí hacia la aurícula derecha por medio del seno coronario, y el menos común es la presencia de una vena cava superior derecha que drena en la aurícula izquierda. Los drenajes anómalos sistémicos, generalmente, se acompañan de otras malformaciones cardiacas (15) y pueden o no hacer parte de síndromes cardioesplénicos, así como algunos reportes que informan una asociación con el síndrome del ojo de gato, síndrome DiGeorge o síndrome velofacial, el cual corresponde a deleción 22q11, también sospechado en este caso (9).
En nuestra paciente se observó el segundo tipo de RVPA, el retorno venoso pulmonar anómalo parcial (RVPAP), el cual representa por sí solo del 0,4 % al 0,7 % de las CC (11,16), con prevalencias en la vida adulta de entre el 0,1 % y el 0,2 % (12). La variante más común (el 60 %) está constituida por un drenaje venoso pulmonar total, en el que hay un tronco venoso único en el pulmón afectado, que se forma por la reunión de las venas pulmonares de origen intersegmentario y drena a alguno de los sitios antes mencionados.
En el caso tratado, la impresión diagnóstica inicial de la paciente se configuró bajo la luz del primer ecocardiograma posnatal, por el signo de la cimitarra, el cual es uno de los componentes del síndrome del pulmón hipogenético, también conocido como síndrome venolobar congénito pulmonar, que se agrupa entre los RVPAP (17). Este presenta conexión de la vena pulmonar derecha anormal con la vena cava inferior (18) y tiene predominio femenino de 2:1 (19). Se asocia con otros defectos cardiacos hasta en un 75 % de los casos (10, 20), entre los que se cuentan la dextroposición, la hipertrofia ventricular, el bloqueo de la rama derecha y la CIA (21), con un alto porcentaje de hipoplasia pulmonar derecha (22). También puede encontrarse secuestro broncopulmonar del lóbulo inferior con displasia de los bronquios derechos superior o medio (21) y suministro arterial sistémico anómalo desde la aorta o una de sus ramas hacia el pulmón derecho (23). Sin embargo, los hallazgos de esta autopsia no configuran un cuadro completo, debido a la conexión anómala de la circulación pulmonar con la aurícula derecha y no con la vena cava inferior.
Aunque a menudo la literatura sobre el tema reporta a los pacientes con RVPAP como asintomáticos y de diagnóstico incidental, si la anomalía afecta más del 50 % del flujo venoso pulmonar, puede volverse clínicamente significativa con cianosis y taquipnea (18), especialmente en pacientes prematuros con cardiopatías de mayor complejidad (6), en los que el pronóstico, a pesar de la disponibilidad de manejo quirúrgico, sigue siendo desfavorable.
Esta paciente, además del RVPAP, presentó otras alteraciones que constituyeron una cardiopatía compleja, por el componente de isomerismo cardiaco, situs inverso parcial, agenesia de los lóbulos pulmonares derechos inferior y medio, poliesplenia, hígado con lóbulos accesorios sésiles bilaterales y lobulación anómala, páncreas dismórfico, útero y anexo derecho dismórficos (útero atrésico, hipoplasia de ovario y agenesia de trompa uterina), frenillo oral, puente nasal ancho, pliegue palmar único izquierdo y pestañas largas. La asociación de otras malformaciones junto con RVPAP es esperada, como se mencionó, en síndromes cardioesplénicos, síndrome del ojo de gato y síndrome DiGeorge-deleción 22q11, como principales síndromes.
Sin embargo, con todas las alteraciones morfológicas vistas en esta paciente, en el momento de la revisión no se encontró en la literatura un síndrome, secuencia o complejo al que pueda corresponder. Casos como el presente ratifican la importancia de trabajo multidisciplinario de los defectos congénitos, ya que varias de las malformaciones descritas no suelen ser captadas en los estudios iconográficos prenatales e, incluso, en los posnatales, pues solo se evidencian en la autopsia. La correcta caracterización de las alteraciones nos acerca a diagnósticos que puedan impactar en la consejería genética.
En conclusión, el RVPA es una entidad poco frecuente, en su mayoría clínicamente leve y también poco diagnosticada, especialmente de forma antenatal; no obstante, puede presentarse acompañado de otras anomalías morfológicas, como en el presente caso, y clínicamente con una cardiopatía compleja con implicaciones mayores en la sobrevida de los pacientes.

