










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Colombiana de Cirugía
versão impressa ISSN 2011-7582versão On-line ISSN 2619-6107
rev. colomb. cir. v.25 n.4 Bogotá out./dez. 2010
(1) Médico, cirujano general, FACS, Departamento de Cirugía, Fundación Santa Fe de Bogotá. Bogotá, D.C., Colombia.
(2) Médico, cirujano general, Departamento de Cirugía, Fundación Santa Fe de Bogotá. Bogotá, D.C., Colombia.
(3) Estudiante, Facultad de Medicina, Universidad de los Andes, Bogotá, D.C., Colombia.
Correspondencia: Manuel Cadena, MD, Bogotá, Colombia. Correo electrónico: manuelcade@gmail.com
Fecha de recibo: 15 de junio de 2010. Fecha de aprobación: 14 de octubre de 2010.
Resumen
Los paragangliomas del órgano de Zuckerkandl son un problema clínico infrecuente. Aunque los pacientes presentan síntomas característicos, como hipertensión arterial de difícil manejo, cefalea y palpitaciones, su diagnóstico se debe considerar de exclusión.
En este artículo se hace una revisión de la literatura y se presenta con caso clínico.
Palabras clave: cuerpos paraaórticos; órgano de Zuckerkandl; paraganglioma; feocromocitoma; hipertensión.
Abstract
Paragangliomas of the Organ of Zuckerkandl constitute an infrequent clinical problem. Although the patients present with the classical triad of hypertension, headaches and palpitations, they should always be considered by exclusion diagnosis. This article includes a literature review and a case presentation.
Key words: para-aortic bodies; organ of Zuckerkandl; paraganglioma; pheochromocytoma; hypertension.
Introducción
El término feocromocitoma proviene de las palabras griegas phaios, chromo y cytoma, las cuales significan oscuro, coloreado y tumor, respectivamente (1,2). El feocromocitoma es un tumor originario de las células cromafines de la cresta neural, que puede desarrollarse en la médula suprarrenal o en los ganglios simpáticos de cualquier parte del cuerpo, y que puede producir catecolaminas (3,4).
Las células cromafines son neuronas simpáticas posganglionares localizadas, principalmente, en la médula suprarrenal; sin embargo, también se han encontrado en tejido suprarrenal accesorio, particularmente en el plexo celiaco (16%) (5). Los tumores de las células cromafines extrasuprarrenales reciben el nombre de paragangliomas, y se pueden encontrar como masas intraabdominales, adyacentes a las glándulas suprarrenales –localizadas en el eje paravertebral y paraaórtico– (85%), o como tumores intratorácicos (15%) o cervicales (1%-3%) (3). Los paragangliomas localizados en la médula suprarrenal se denominan feocromocitomas (6,7).
De acuerdo con lo anterior, la Organización Mundial de la Salud (OMS) define como feocromocitoma al “tumor de células cromafines de la médula suprarrenal”, y como “paragangliomas extrasuprarrenales”, a todos los demás tumores relacionados (con una localización y tipo determinados) (8-10). Más del 90% de ellos se originan en la médula suprarrenal mientras que, aproximadamente, 10% son extrasuprarrenales (3). Según se ha descrito, el porcentaje de distribución de los feocromocitomas es el siguiente: 10% son extrasuprarrenales, 10% son familiares, 10% son bilaterales y 10% son malignos (11-14).
Los paragangliomas extrasuprarrenales pueden originarse, hasta en 20% de los casos, en el órgano de Zuckerkandl (3,5). Este órgano fue descrito por Emil Zuckerkandl (1901) como agrupaciones de paraganglios de color café claro, encapsulados y unidos por un istmo a la superficie anterior de la aorta, justo entre el origen de la arteria mesentérica inferior y la bifurcación de las arterias iliacas, y formados por tejido de células cromafines (3,15). Para el 2006, se habían reportado en la literatura unos 135 casos de tumores del órgano de Zuckerkandl (14).
Cabe anotar que los feocromocitomas son tumores raros, con una incidencia anual de 1 a 4 por 1’000.000, de los cuales, 0,5% se presentan con hipertensión, y 4% con una masa de hallazgo incidental o “incidentaloma” (16). El tiempo que transcurre desde el inicio de los síntomas hasta que se hace el diagnóstico es de tres años, aproximadamente; la edad promedio de diagnóstico está entre los 40 y los 50 años, y la incidencia es similar entre hombres y mujeres (13,16). En la mayoría de los casos los tumores son esporádicos, solitarios y de origen suprarrenal (16).
No existen criterios establecidos para definir su carácter maligno; sin embargo, se considera que la invasión capsular, una masa mayor de 5 cm, un peso mayor de 80 g y la recurrencia o presencia de enfermedad metastásica lo indican (1,8,10,17-20). En este último aspecto, los feocromocitomas difieren en gran medida de los paragangliomas: mientras que para los primeros la prevalencia de enfermedad metastásica es de 5%, para los paragangliomas es de 33% en el momento del diagnóstico (1,9,16).
Diagnóstico
La tríada sintomática de taquicardia, diaforesis y cefalea, que se encuentra presente en 40% a 80% de los pacientes, es muy sensible y específica para el diagnóstico (1,10,14,15,20,21). Las manifestaciones clínicas relacionadas con los tumores del tejido cromafín dependen de la secreción de catecolaminas, y su patrón dominante es principalmente la hipertensión, paroxística o sostenida, por niveles elevados de noradrenalina (1,16,20-22).
Otros síntomas menos frecuentes son: palidez, sensación de pánico, náuseas, fiebre, rubor, pérdida de peso (23) y, debido al bloqueo de la liberación de insulina causado por las catecolaminas, también se puede encontrar diabetes mellitus (24).
Los feocromocitomas pueden presentarse como parte de síndromes hereditarios o en forma esporádica (16,25,26). Los feocromocitomas hereditarios se encuentran en el síndrome de neoplasia endocrina múltiple de tipo 2 (Multiple Endocrine Neoplasia 2, MEN 2), en el síndrome de Hippel-Lindau, en la neurofibromatosis de tipo 1 y en los paragangliomas familiares, entre otros (22,27). Las formas esporádicas se hallan usualmente entre los 40 y los 50 años de edad, mientras que las formas hereditarias a edades más tempranas (1,9,22,28).
Aunque su aparición es rara, dentro del diagnóstico diferencial de un paciente con hipertensión crónica de difícil manejo es indispensable que se tengan en cuenta estas enfermedades (14,16).
El diagnóstico se hace principalmente mediante la clínica y el estudio bioquímico (29). En primer lugar, se tiene la sospecha clínica, que se confirma por medio de la medición de los niveles de las catecolaminas y sus metabolitos en orina de 24 horas que, en caso afirmativo, resultan con excreción aumentada (30,31). El examen más sensible para la evaluación de los pacientes con sospecha de feocromocitoma es la medición de metanefrinas libres en plasma, con una sensibilidad del 99% (22).
Finalmente, la masa puede ser localizada mediante tomografía computadorizada (TC) y resonancia magnética (RM); o con gammagrafía con metayodo bencilguanidina (MIBG) (27,32-35). Las dos primeras permiten una adecuada localización de la masa, ya que aportan una gran resolución espacial; sin embargo, su especificidad es limitada (16,31). La gammagrafía con MIBG es una excelente herramienta para el diagnóstico del feocromocitoma si se tiene en cuenta que esta sustancia se concentra en tejidos simpatoadrenérgicos, especialmente en el tejido cromafín, y que puede ser marcada con isótopos de yodo, que ponen en evidencia la localización del tumor (35).
Entre los agentes utilizados para los estudios de imaginología, la 123I-metayodobencilguanidina (123I-MIBG) ha demostrado superioridad respecto a la 131I-metayodobencilguanidina (131I-MIBG) por las siguientes consideraciones: la 131I-metayodobencilguanidina tiene una vida media de 8,2 días, mientras que, para la 123I-metayodobencilguanidina, es tan sólo de 13,2 horas. Además, esta última genera una dosis mayor de rayos gamma que le confieren una mayor especificidad y, por lo tanto, una mejor imagen (35).
Asimismo, se ha encontrado que la sensibilidad y la especificidad de la 131I-MIBG para localizar feocromocitomas suprarrenales es de 77% a 90%; y de 95% a 100% para los extrasuprarrenales; y la de la 123I-MIBG es de 83% al 100%, y de 95% a 100%, respectivamente (35). Estudios más recientes demuestran que la sensibilidad y la especificidad de la 123I-MIBG son de 91,5% y 100%, con valor diagnóstico positivo de 100% y valor diagnóstico negativo de 83%.
No obstante, van Der Harst (2006) reportó que este estudio de imaginología tiene una menor sensibilidad para la detección de paragangliomas familiares (MEN2A/2B), bilaterales, metástasis y localización extrasuprarrenal (36). Asimismo, van Der Horst-Schrivers (2006) reportó que la sensibilidad y especificidad para la detección de tumores en la glándula suprarrenal son del 98%, y que la sensibilidad para la detección de tumores extrasuprarrenales y malignidad es de 98% y 79% respectivamente (37). Bhatia (2008) estableció, que con 123I-MIBG la detección de feocromocitomas es de 85% y la de paragangliomas es de 58% (35).
Aunque, en general, la exactitud diagnóstica de la gammagrafía con MIBG es menor en tumores extrasuprarrenales y en paragangliomas malignos; éste continúa siendo el examen de elección en el diagnóstico de feocromocitomas y paragangliomas. Nuevas investigaciones apuntan hacia el uso de tomografías por emisión de positrones con 6-[18F] fluorodopamina, que podrían llegar a remplazar los estudios con MIBG; sin embargo, no han sido concluyentes (35).
Tratamiento
Es importante tener en cuenta que el manejo interdisciplinario es indispensable para el diagnóstico y tratamiento de esta enfermedad. Sin embargo, en esta revisión se profundiza en los tratamientos prequirúrgico y quirúrgico de elección. El primero tiene como objeto disminuir la morbimortalidad asociada a los altos niveles de catecolaminas a que se expone el paciente durante el procedimiento quirúrgico, al igual que a los riesgos intrínsecos a la intervención; y el segundo, el fin de realizar la resección completa de la masa (22,33).
Para el manejo quirúrgico se puede utilizar la laparoscopia o la cirugía convencional; sin embargo, la elección debe guiarse por la adecuada selección del paciente (16,38-42).
Actualmente, y de acuerdo con estudios recientes, la cirugía por laparoscopia se considera como el método de elección, tanto para los feocromocitomas suprarrenales como para los extrasuprarrenales, pues reduce significativamente la morbilidad, la estancia hospitalaria y la cantidad de analgesia en el periodo posoperatorio, comparativamente con el procedimiento convencional (14,16,33,43-45).
No obstante, aquellos casos en los cuales el tumor es mayor de 6 cm y se encuentra muy adherido a las estructuras adyacentes, como sucede con los paragangliomas extrasuprarrenales retroperitoneales, se prefiere usar la cirugía convencional, y la lumbotomía es el método con mejores resultados (18,33,46,47).
En los casos con metástasis o con bordes quirúrgicos comprometidos, se puede utilizar radioterapia o quimioterapia, pero sólo con intención paliativa (35,47-49).
Presentación de caso
El paciente es un hombre de 38 años de edad que presenta un cuadro clínico de cuatro años de evolución, consistente en episodios súbitos de cefalea global, palpitaciones y diaforesis. Por este motivo, consultó en múltiples ocasiones al servicio de urgencias de diferentes instituciones, donde le encontraron cifras de tensión arterial hasta de 240/160 mm Hg. Fue estudiado por el Servicio de Cardiología; sin embargo, no fue posible establecer la causa del cuadro clínico.
En enero de 2007, presentó una nueva crisis hipertensiva acompañada de dolor precordial, por lo cual se decidió practicar un ecocardiograma de estrés con dobutamina, que debió ser interrumpido por presentar episodio hipertensivo. Por no encontrar hallazgos relevantes, se decidió continuar con el manejo farmacológico.
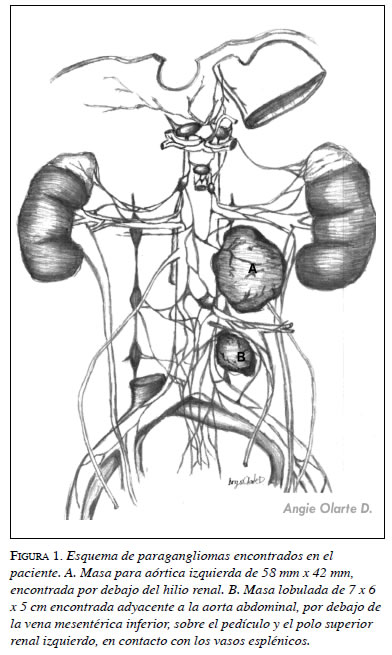
Debido a la persistencia de las crisis y con base en el diagnóstico de paciente hipertenso crónico de difícil manejo, se decidió hacer un análisis riguroso. En el estudio detallado, se solicitaron niveles de catecolaminas, metanefrinas (tabla 1), gammagrafía con MIBG y RM, que arrojaron resultados concluyentes: se encontró aumento de los niveles de ácido vanilmandélico y una masa paraaórtica izquierda de 58 por 42 mm, situada por debajo del hilio renal. Esto permitió hacer el diagnóstico de paraganglioma del órgano de Zuckerkandl (figura 1).
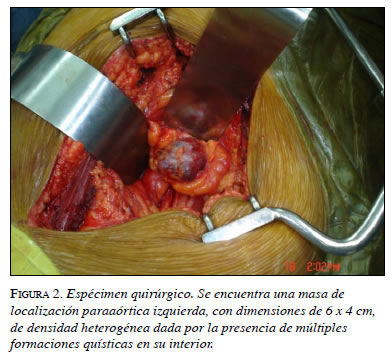
Por consiguiente, se le practicó resección completa de la masa descrita en abril de 2008 (figura 2). Durante la intervención, el paciente presentó cifras elevadas de tensión arterial (máxima de 253/135 mm Hg), e importante hipovolemia que se manejó con múltiples transfusiones. No obstante, su evolución fue satisfactoria y dos días después desaparecieron los síntomas y se normalizó la tensión arterial.
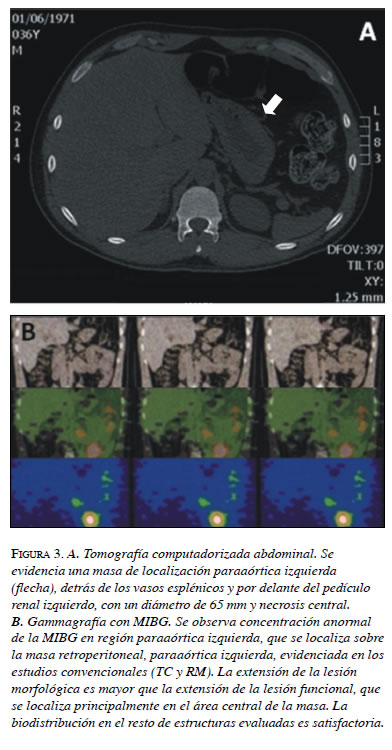
En agosto de 2008, reaparecieron los síntomas, acompañados de nuevas crisis hipertensivas, por lo cual se decidió determinar los niveles de ácido vanilmandélico (tabla 1) y practicar una TC, en la que se encontró una lesión residual de localización paraaórtica, detrás de los vasos esplénicos y por delante del pedículo renal izquierdo, con un diámetro de 65 mm y necrosis central (figura 3). A partir de este hallazgo se decidió programarlo nuevamente para cirugía. En el periodo posoperatorio, el electrocardiograma presentó inversión de la onda T en dos derivaciones y trastornos en la repolarización, sin síntomas asociados.
Discusión
Manejo preoperatorio
Las catecolaminas liberadas por la manipulación del tumor durante la operación pueden ocasionar complicaciones serias como crisis hipertensivas, arritmias cardíacas, edema pulmonar e isquemia del miocardio (50-52). La administración de medicamentos antes de la cirugía se origina de este supuesto, y busca reducir la morbimortalidad asociada (53). Se recomienda que este manejo sea realizado en un periodo entre 10 días y 2 semanas antes de la intervención (16).
Asimismo, el paciente con un feocromocitoma o un paraganglioma puede tener disminución de volumen debido a la vasoconstricción crónica que genera la enfermedad; además, en el periodo perioperatorio puede sufrir alteraciones de su equilibrio hidroelectrolítico, por lo cual, antes de llevarlo a cirugía, se debe corregir la pérdida asociada de volemia (16,54). Sin embargo, existe controversia en la literatura sobre esta medida; por ejemplo, el grupo de la Clínica Cleveland no utiliza rutinariamente antagonistas adrenérgicos en el periodo previo a la cirugía (5), pues no hay pruebas suficientes de que esta medida reduzca las complicaciones asociadas (53).
Por otra parte, el bloqueo de los receptores adrenérgicos puede exacerbar la hipotensión posoperatoria y privar al cirujano de la hipertensión como signo de alarma, ya que puede indicarle la presencia de otro tumor oculto (12,27,53). No obstante, los investigadores del grupo de la Clínica Lahey los utilizan en casos seleccionados, principalmente en aquellos pacientes que presentan hipertensión grave, signos de considerable disminución de volumen y crisis hipertensivas no controladas, graves y frecuentes (5,26).
Si se decide utilizar el método tradicional (premedicación con antagonistas adrenérgicos), se debe iniciar con alfa-bloqueadores (53,55), como son la fenoxibenzamina, la fentolamina y la prazosina. Entre estos, la fenoxibenzamina se considera el de primera elección (5,22,27) pues, en comparación con los bloqueadores competitivos, logra un adecuado control de la presión arterial intraoperatoria al no ser desplazada en presencia de un exceso de catecolaminas. Sin embargo, se puede presentar un mayor riesgo de episodios hipotensivos durante el periodo posoperatorio (16,56).
Otras alternativas terapéuticas son los betabloqueadores, como el labetalol o las dihidropiridinas (bloqueadores de canales de calcio), utilizadas como monoterapia o en combinación con antagonistas alfa-adrenérgicos (11,57).
El manejo preoperatorio puede generar efectos de ortostatismo y taquicardia refleja debido a la vasoconstricción (53,54). Esta última puede tratarse con betabloqueadores, que no deben administrarse antes de los alfa-bloqueadores, debido a que el bloqueo beta puede inhibir la vasodilatación inducida por la epinefrina y, así, llevar a una hipertensión más significativa, con falla cardiaca y edema pulmonar (27,54).
Manejo quirúrgico
Como lo menciona Rabii, “desde su descripción por Gagner y colaboradores (1992), la suprarrenalectomía laparoscópica se ha considerado como método de referencia para el manejo quirúrgico en enfermedad suprarrenal benigna” (58), pues ofrece ventajas en comparación con la cirugía abierta, como son: menor periodo de estancia hospitalaria, reducción de las complicaciones intraoperatorias y disminución del dolor posoperatorio (43).
Tradicionalmente, se ha pensado que el abordaje laparoscópico es una contraindicación relativa en el caso de los feocromocitomas, debido principalmente a la incertidumbre en cuanto a su carácter maligno y las posibles complicaciones hemodinámicas intraoperatorias que pueden presentarse (29,59). Sin embargo, se ha encontrado que la incidencia de complicaciones hemodinámicas durante la suprarrenalectomía, laparoscópica o abierta, son similares (18,40,43,44,60).
Para los dos casos, las principales complicaciones se deben a grandes cambios de la tensión arterial durante la manipulación de la masa, o se generan por el neumoperitoneo causado por la insuflación de dióxido de carbono (CO2) (21,53), y pueden presentarse a pesar del manejo preoperatorio con alfa-bloqueadores y beta-bloqueadores. También, se ha reportado hemorragia intraoperatoria por lesión vascular (56,61).
La elección ideal para el manejo quirúrgico de los feocromocitomas y los paragangliomas es controvertida actualmente, ya que los diversos estudios que se han realizado tienen limitaciones generadas por el tamaño de la muestra (59) y por el bajo reporte de casos sobre su abordaje (39). Algunos autores sugieren laparoscopia transperitoneal para los feocromocitomas bilaterales o los paragangliomas (24); sin embargo, se han reportado casos en los que un abordaje laparoscópico retroperitoneal inicial tuvo que convertirse a laparotomía, y en todos los casos los tumores fueron mayores de 5,5 cm de diámetro (57).
La recomendación de nuestro Servicio de Cirugía es que, en casos de tumores grandes (más de 6 cm), adherentes, hipervasculares, con extensión retrocava o signos de neoplasia maligna en las imágenes, se debe optar por un abordaje abierto que permita un adecuado control vascular y una resección completa (40).
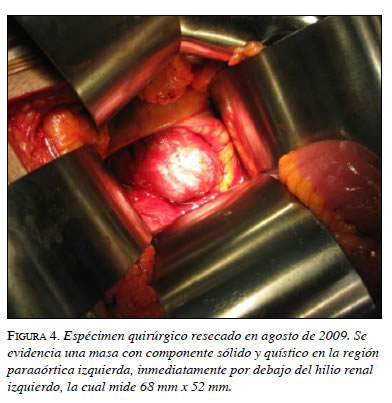
En el paciente que presentamos aquí, y de acuerdo con la información obtenida y las características anatomopatológicas encontradas, se decidió realizar un abordaje abierto (figura 4).
Anatomía patológica
Descripción macroscópica del espécimen
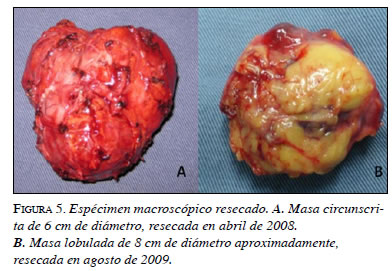
En la segunda intervención se encontró una masa lobulada con un tamaño de 7 x 6 x 5 cm, con un peso de 75 g, adyacente a la aorta abdominal, por debajo de la vena mesentérica inferior, sobre el pedículo y el polo superior renal izquierdo, y en contacto con los vasos esplénicos (figura 5). En el corte macroscópico, se observó un parénquima homogéneo de color rojizo con algunas áreas quísticas. Además, el tejido mostraba cambios por lisis a nivel central.
Citología e inmunohistoquímica
El avance de la biología molecular ha permitido un incremento en la disponibilidad de las técnicas diagnósticas para identificar la histopatología de los tumores endocrinos de forma más precisa, específicamente el inmunofenotipo y el genotipo tumoral (1,16). A pesar de esto, la tipificación, tanto del paraganglioma como del feocromocitoma respecto a su potencial maligno, es difícil (1,46), ya que el único criterio absoluto para determinar dicho potencial es la presencia de metástasis del tejido cromafín en sitios en donde no se esperaría encontrarlo (1). Por esta razón, es importante tener en cuenta que existe la posibilidad de que el tumor presente un gran crecimiento local, sin que esto se correlacione con su potencial metastásico (39).
Las metástasis de los feocromocitomas son raras y se encuentran en 5% de los casos en el momento del diagnóstico. En los paragangliomas mayores de 5 cm se encuentran hasta en el 33% (8,9,11,62). Los lugares más frecuentes de metástasis son el hueso, el pulmón, el hígado y los ganglios linfáticos (13,16).
El ejercicio diagnóstico se basa principalmente en encontrar la mayor cantidad de factores clínicos, imaginológicos, citológicos e inmunohistoquímicos para determinar el potencial maligno del tumor. Para esto se han propuesto múltiples escalas que dan una aproximación al potencial maligno. Por ejemplo, Thompson (2002) propuso el sistema de puntuación PASS (Pheochromocytoma of the Adrenal Gland Scaled Score), específico para feocromocitomas, y que se basa en un puntaje máximo de 20 características histológicas, que incluyen, entre otras, arquitectura celular, tamaño celular, invasión y actividad mitótica (49). Generalmente, los puntajes PASS mayores de 4 evidencian asociación con presencia de metástasis. No obstante, los puntajes inferiores a 4 no descartan la posibilidad de su desarrollo (49).
La inmunohistoquímica ha producido resultados muy variables respecto a la determinación del carácter maligno tumoral (1,46). La literatura reporta diferentes moléculas y péptidos expresados por los tumores, benignos y malignos, pero los datos no son constantes (16). La lista de los marcadores de inmunohistoquímica correlacionados con el potencial maligno es amplia, e incluye: la transcriptasa inversa de la telomerasa (telomerase reverse transcriptase), la proteína 90 de choque de calor, el factor de crecimiento vascular endotelial, el factor 1 de la transcripción inducible por hipoxia, la ciclooxigenasa, la cadherina N, la tenascina C y productos de escisión de la cromogranina y la secretogranina. Sin embargo, ninguno de ellos ha sido completamente validado y no se utilizan en la práctica diagnóstica (20,32,63,64). Asimismo, las pruebas moleculares, como el índice de proliferación celular Ki 67 y la proteína S100 que identifica células sustentaculares, no han mostrado resultados concluyentes en los reportes de la literatura (1,10,32).
En la panorámica de la masa resecada (figura 6a) se observa el margen –en tinta negra–, indicador del límite del compromiso de la lesión que, en este caso, no presentaba invasión de otras estructuras. Además, se encuentra una arquitectura mixta entre un patrón alveolar (Zellballen) y otro trabecular, usualmente encontrada en los feocromocitomas (figura 6b).
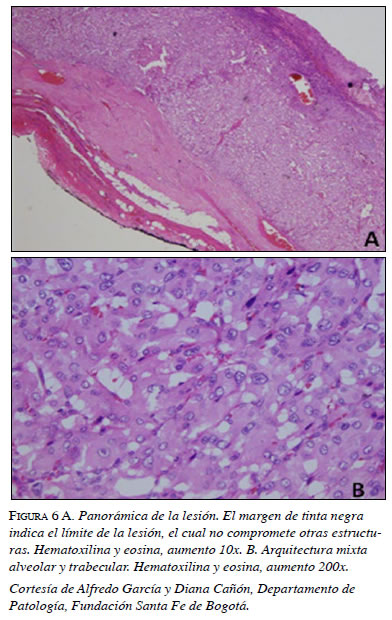
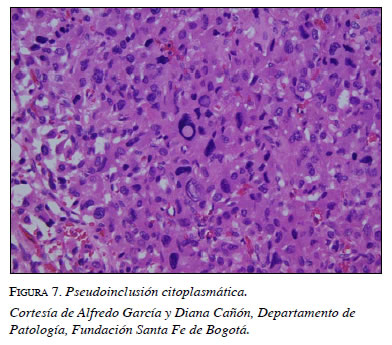
En la citología se encuentra pleomorfismo nuclear, citoplasma granular y basofílico abundante, características de las células cromafines (figura 6b). En el examen histopatológico se encuentran inclusiones globulares hialinas del citoplasma, hallazgo característico reportado por la literatura (figura 7).
Los estudios de inmunoperoxidasa, por su parte, muestran reacción en las células tumorales para la cromogranina A (CGA): esta prueba es importante para diferenciar las neoplasias suprarrenales corticales, los feocromocitomas y los tumores metastásicos de otros tipos diferentes a los de origen neuroendocrino (figura 8a).
Focalmente y por medio de la proteína S100, se identifican células sustentaculares alrededor de los nidos de células (organización alveolar) (figura 8b). Aunque esta prueba no se realiza rutinariamente en los protocolos, es útil para la confirmación del tumor como origen de la secreción hormonal y para determinar su carácter maligno (1,8). El índice de proliferación celular Ki 67 fue menor de 1% (figura 8c).
De acuerdo con esto, los hallazgos morfológicos y de inmunofenotipo corresponden a un feocromocitoma benigno. En este caso, al tratarse de un tumor extrasuprarrenal, los hallazgos corresponden al diagnóstico hecho de paraganglioma del órgano de Zuckerkandl, probablemente benigno.
Periodo posoperatorio
El tejido cromafín de los feocromocitomas, derivado de la cresta neural, produce secreción excesiva de catecolaminas, que tiene consecuencias a distancia (52); clásicamente, éstas se manifiestan con la tríada mencionada anteriormente. Sin embargo, se ha encontrado que también pueden asociarse con miocarditis aguda, infarto agudo del miocardio, cardiomiopatías y colapso hemodinámico (26,50).
Cuando el paciente sufre un infarto agudo del miocardio, el electrocardiograma puede mostrar elevaciones en el segmento ST, aunque comúnmente no es así (65). La mayoría de estos pacientes no tiene aterosclerosis coronaria significativa, a diferencia de lo que ocurre en la presentación clásica del infarto agudo del miocardio (53). Esto puede deberse a los altos niveles de catecolaminas liberadas por el tumor, que aumentan el metabolismo del miocardio y producen espasmo de las arterias coronarias, lo que desencadena angina, infarto y cardiomiopatía (51). Los pacientes con feocromocitomas también pueden presentar taquicardias supraventriculares y, con menor frecuencia, taquicardia ventricular, taquicardia ventricular sostenida (torsades de pointes) y fibrilación ventricular (52), así como, arritmias y falla cardiaca (51,61).
Estas respuestas hemodinámicas variables son atribuidas a la modulación cardiaca simpático-vagal, que se pueden correlacionar con las concentraciones plasmáticas de norepinefrina (13,52). Se ha demostrado que en estos pacientes se pueden alterar sus variables fisiológicas por la liberación de catecolaminas durante la resección del tumor (13). De la misma forma, los altos niveles plasmáticos de catecolaminas pueden aumentar directamente la tensión arterial durante el procedimiento (50), como sucedió con el paciente que presentamos aquí.
La aparición de cambios isquémicos en el electrocardiograma puede ser el resultado de la estimulación miocárdica por niveles elevados de catecolaminas; generalmente, es transitoria y sugiere miocarditis tóxica, más que un infarto transmural verdadero (65).
En el periodo posoperatorio, el electrocardiograma de este paciente mostró inversión de la onda T en la pared anterolateral (DI y AVL), con posterior alteración de la repolarización en V4; esto se interpretó inicialmente como un posible trastorno secundario a la sobrecarga ventricular izquierda debida a las crisis hipertensivas transitorias durante el procedimiento. Por este motivo, se decidió solicitar niveles séricos de troponina, la que resultó positiva (0,305 ng/mL). Este hallazgo se interpretó como el resultado de un evento isquémico posoperatorio: infarto agudo del miocardio sin elevación del ST de tipo 2.
Más tarde, se hizo un control de troponina, que mostró disminución respecto al valor previo (0,1 ng/mL), lo que descartó un evento coronario en curso. En el ecocardiograma transesofágico se encontró hipertrofia concéntrica leve del ventrículo izquierdo, con trastornos de la contractilidad segmentaria, y disfunción diastólica estadio I, hallazgos que pueden sugerir una cardiomiopatía inducida por catecolaminas, dependientes del feocromocitoma. La recuperación de la función cardiaca de la disfunción ventricular, que puede llegar a ser grave después de la remoción del tumor, es la evidencia indirecta de que el corazón fue expuesto a altas tensiones arteriales por un tiempo prolongado.
Pruebas genéticas
Síndromes hereditarios
Cuando se encuentra un tumor endocrino de origen suprarrenal o extrasuprarrenal, se debe considerar la pertinencia de practicar estudios para síndromes hereditarios. Existen dos razones importantes por las cuales se deben practicar estos estudios a todos los pacientes con tumores de esta clase. La primera es que se sabe que las formas hereditarias de estos tumores se encuentran asociadas a otras neoplasias (6,14,66,67); por lo tanto, hacer el diagnóstico temprano permite el tratamiento oportuno, la vigilancia precoz del paciente (16,26) y, además, hacer la tamización de los familiares. La segunda es que los pacientes con mutaciones germinales tienen un riesgo mayor de presentar tumores múltiples muy recurrentes, por lo que su estudio permite el seguimiento y la vigilancia continua del paciente, para la detección oportuna de neoplasias malignas (16,26).
Todos los pacientes con feocromocitoma o paraganglioma, con historia familiar o características clínicas sugestivas de un síndrome hereditario, deben tener estudio genético (25,68). Las características clínicas que pueden sugerir la presencia de un feocromocitoma hereditario son: muerte súbita a edad temprana, hipertensión o accidente cerebrovascular en jóvenes o durante el embarazo, y respuesta hipertensiva a la anestesia (68). Además de éstas, se debe considerar que los pacientes con tumores bilaterales o multicéntricos tienen una alta probabilidad de presentar síndromes hereditarios de paraganglioma o de feocromocitoma (68).
De la misma forma, deben estudiarse los pacientes con paragangliomas simpáticos, especialmente en presencia de tumores múltiples (16,26,68). El perfil bioquímico del paciente puede orientar hacia el tipo de mutación germinal: cuando el perfil bioquímico muestra elevación de los valores de epinefrina y metanefrina, el gen RET debe ser estudiado en primera instancia; mientras que, si el perfil bioquímico muestra elevación de la norepinefrina y la normetanefrina, el gen VHL debe ser estudiado en primera instancia; y si los genes RET y VHL son negativos, deben analizarse los genes SDHB y SDHD (68).
Los pacientes cuya edad de inicio de la enfermedad es anterior a los 20 años deben estudiarse (25,68), ya que algunos estudios reportan su asociación con enfermedad hereditaria, principalmente con el síndrome de von Hippel-Lindau (66,69). Por el contrario, en pacientes mayores de 50 años que presentan paraganglioma o feocromocitoma de presentación esporádica, la probabilidad de presentar mutaciones de los genes VHL, MEN2, SDHB o SDHD es de 1,3%, lo que hace innecesaria la práctica de estudios genéticos para este grupo de pacientes (25,68,70).
En este reporte, al paciente no se le realizaron estudios genéticos, ya que se consideró que no existían hallazgos sugestivos o indicios en su historia familiar o personal de presencia de otros tumores o de mutaciones germinales.
Recurrencia y seguimiento
Aunque se intervenga quirúrgicamente y con intención curativa, un paciente con feocromocitoma o paraganglioma, siempre puede enfrentarse al riesgo de recurrencia del tumor (8). En los estudios de casos en serie, y con el análisis de factores diversos como la localización del tumor, el número de masas, su tamaño, sus características histopatológicas y la existencia de enfermedad hereditaria, se ha demostrado la variabilidad de la recurrencia de esta enfermedad (34,62,71,72). Esto ha permitido hacer aproximaciones prospectivas respecto al posible comportamiento de los tumores, según el contexto del paciente y en términos de recurrencia de la enfermedad (72): los pacientes que presentan recurrencia tienden a ser más jóvenes, y a tener tumores significativamente más grandes y más pesados en la primera intervención quirúrgica (72).
Hay estudios que han reportado recurrencias de 33% en pacientes con tumores extrasuprarrenales, en comparación con 14% en pacientes con tumores suprarrenales (16,22). También, se ha encontrado que es 33% más frecuente si hay enfermedad familiar, en comparación con 13% en los casos que se presentan esporádicamente (16).
El pronóstico a largo plazo para los pacientes, después de la resección de un tumor solitario esporádico, es bueno (22). Sin embargo, la hipertensión puede persistir después de la cirugía, aproximadamente, en 50% de los pacientes (16,26 ). Es por esta razón que todos los pacientes deben ser seguidos anualmente, por un total de 10 años; y en los casos de los pacientes con tumores extrasuprarrenales, o de feocromocitoma o paraganglioma familiar, el seguimiento debe ser indefinido (16,72).
Esto coincide con la presentación recurrente de paragangliomas en nuestro paciente, pues a pesar de ser esporádico y sin presencia de antecedentes familiares, al ser un tumor extrasuprarrenal se aumenta la incidencia de presentación. De acuerdo con los hallazgos y la conducta seleccionada, el seguimiento en este caso debe ser permanente, a pesar de tener un buen pronóstico a largo plazo.
Conclusión
En un hombre de 38 años de edad, con historia de cuatro años de crisis hipertensivas de difícil manejo, palpitaciones, cefalea y diaforesis, sin historia familiar ni personal asociada, se hizo diagnóstico de feocromocitoma extrasuprarrenal del órgano de Zuckerkandl o paraganglioma.
En la primera intervención quirúrgica se le extrajo una masa paraaórtica. Un año después había recurrencia de la enfermedad y se le encontró una nueva masa, con características histopatológicas que confirmaron el diagnóstico anterior e indicaron un carácter benigno.
Se concluye que es indispensable el manejo preoperatorio para reducir la morbimortalidad asociada. Entre las complicaciones asociadas al tratamiento quirúrgico se encuentran crisis hipertensivas, arritmias cardiacas, edema pulmonar e isquemia miocárdica. Se recomienda que el manejo preoperatorio se realice entre los 10 días y las dos semanas previas a la intervención, y de acuerdo con las condiciones y antecedentes del paciente.
La suprarrenalectomía laparoscópica se ha considerado como el método de referencia para el manejo quirúrgico en enfermedad suprarrenal benigna. No obstante, algunos autores reportan que este abordaje es una contraindicación relativa en el caso de los feocromocitomas, debido a la incertidumbre en cuanto a su carácter maligno y las posibles complicaciones hemodinámicas intraoperatorias. Sin embargo, se ha encontrado que la incidencia de complicaciones hemodinámicas durante la suprarrenalectomía laparoscópica o el procedimiento quirúrgico abierto son similares.
Nuestro Servicio de Cirugía sugiere la práctica de laparoscopia transperitoneal para el tratamiento quirúrgico de feocromocitomas bilaterales o de paragangliomas, mientras que, en los casos de tumores grandes (mayor de 6 cm), que son adherentes, hipervasculares, con extensión al espacio posterior a la vena cava, o que muestran signos de ser malignos en las imágenes, se recomienda un abordaje abierto que permita un adecuado control vascular y una resección completa.
La liberación excesiva de catecolaminas por el tumor durante la manipulación quirúrgica puede producir hipertensión, arritmia, hipertrofia miocárdica y falla cardiaca, así como las otras manifestaciones mencionadas.
No existen criterios establecidos para definir el carácter maligno de estos tumores; sin embargo, se consideran factores indicativos la invasión capsular, una masa mayor de 5 cm, un peso superior a 80 g, y recurrencia o presencia concomitante de enfermedad metastásica. Esta última ha demostrado ser el único criterio absoluto, y los lugares donde más frecuentemente aparecen dichas metástasis son el hueso, el pulmón, el hígado y los ganglios linfáticos.
Los feocromocitomas o los paragangliomas hereditarios se presentan en la neoplasia endocrina múltiple de tipo 2 (MEN 2), en el síndrome de von Hippel-Lindau, en la neurofibromatosis de tipo 1 y en los paragangliomas familiares. Las formas esporádicas de feocromocitoma usualmente se encuentran en pacientes cuya edad está entre los 40 y los 50 años; mientras que las formas hereditarias se presentan en pacientes de edades más tempranas.
El estudio genético debe practicarse en todos los pacientes que posean las características mencionadas (antecedentes familiares, presencia de otros tumores o mutaciones germinales, entre otros), y éste puede orientarse según el perfil bioquímico del paciente.
La recurrencia tiende a presentarse en pacientes jóvenes con tumores extrasuprarrenales, de localización derecha, significativamente grandes y pesados, y con historia de enfermedad familiar hereditaria.
Finalmente, el seguimiento para todos los pacientes debe ser anual, durante 10 años; y para los pacientes con tumores extrasuprarrenales o síndromes familiares, debe ser indefinido.
Es indispensable considerar el manejo interdisciplinario para hacer la aproximación diagnóstica y terapéutica de estos pacientes, en primer lugar, mediante un diagnóstico clínico, bioquímico e imaginológico y, en segundo lugar, mediante un adecuado manejo antes, durante y después de la cirugía. Éste es un diagnóstico que se debe considerar en todo paciente con hipertensión crónica de difícil manejo y sin causa conocida.
Agradecimientos
A los doctores Mario Ruiz, Alfredo García, Diana Cañón y Gonzalo Ucrós, de la Fundación Santa Fe de Bogotá.
A los departamentos de Cirugía, Anestesiología, Medicina Interna, Imágenes Diagnósticas y Medicina Nuclear, y Patología del Hospital Universitario, Fundación Santa Fe de Bogotá.
Referencias
1. McNicol AM. Histopathology and immunohistochemistry of adrenal medullary tumors and paragangliomas. Endocr Pathol. 2006;17:329-36. [ Links ]
2. Kantorovich V, Pacak K. Pheochromocytoma and paraganglioma. Prog Brain Res. 2010;182:343-73. [ Links ]
3. Glenn F, Gray GF. Functional tumors of the organ of Zuckerkandl. Ann Surg. 1976;183:578-86. [ Links ]
4. Roman S. Pheochromocytoma and functional paraganglioma. Curr Opin Oncol. 2004;16:8-12. [ Links ]
5. Subramanian A, Maker VK. Organs of Zuckerkandl: Their surgical significance and a review of a century of literature. Am J Surg. 2006;192:224-34. [ Links ]
6. Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91:827-36. [ Links ]
7. Karagiannis A, Mikhailidis DP, Athyros VG, Harsoulis F. Pheochromocytoma: An update on genetics and management. Endocr Relat Cancer. 2007;14:935-56. [ Links ]
8. Tischler AS, Kimura N, McNicol AM. Pathology of pheochromocytoma and extra-adrenal paraganglioma. Ann N Y Acad Sci. 2006;1073:557-70. [ Links ]
9. McNichol AM. Differential diagnosis of pheochromocytomas and paragangliomas. Endocr Pathol. 2001;12:407-15. [ Links ]
10. McNicol AM. Update on tumours of the adrenal cortex, phaeochromocytoma and extra-adrenal paraganglioma. Histopathology. 2010 Aug 16. [Epub ahead of print]. [ Links ]
11. Tischler AS. Pheochromocytoma and extra-adrenal paraganglioma: updates. Arch Pathol Lab Med. 2008;132:1272-84. [ Links ]
12. Álvarez TR, Álvarez T, Portela O, Olvera BC, Burgos ZA. Feocromocitoma. Presentación de un caso y revisión de la literatura. Rev Mex Cir Endoscop. 2007;8:148-56. [ Links ]
13. Álvarez R. Feocromocitoma. Presentación de un caso y revisión de la literatura. Asociación Mexicana de Cirugía Endoscópica, AC. 2007;8:148-56. [ Links ]
14. Arroyo-Martínez L, Álvarez-Pertuz H, Acuña-Calvo J, Montoya-Calles J. Paraganglioma funcional extra-adrenal. Acta Méd Costarric. 2006;48:39-42. [ Links ]
15. Hayes WS, Davidson AJ, Grimley PM, Hartman DS. Extraadrenal retroperitoneal paraganglioma: Clinical, pathologic, and CT findings. AJR Am J Roentgenol. 1990;155:1247-50. [ Links ]
16. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366:665-75. [ Links ]
17. Adler JT, Meyer-Rochow GY, Chen H, Benn DE, Robinson BG, Sippel RS, et al. Pheochromocytoma: Current approaches and future directions. Oncologist. 2008;13:779-93. [ Links ]
18. Liao CH, Chueh SC, Lai MK, Hsiao PJ, Chen J. Laparoscopic adrenalectomy for potentially malignant adrenal tumors greater than 5 centimeters. J Clin Endocrinol Metab. 2006;91:3080-3. [ Links ]
19. Portela-Gomes GM, Stridsberg M, Grimelius L, Falkmer UG, Falkmer S. Expression of chromogranins A, B, and C secretogranin II) in human adrenal medulla and in benign and malignant pheochromocytomas An immunohistochemical study with region-specific antibodies. APMIS. 2004;112:663-73. [ Links ]
20. Elder EE, Xu D, Hoog A, Enberg U, Hou M, Pisa P, et al. KI-67 and hTERT expression can aid in the distinction between malignant and benign pheochromocytoma and paraganglioma. Mod Pathol. 2003;16:246-55. [ Links ]
21. Sotolongo Y, Vigoa L, Pérez J. Feocromocitoma y anestesia: revisión del tema a propósito de un caso. Rev Cubana Cir [online]. 2002;414). Disponible en http://bvs.sld.cu/revistas/cir/vol41_4_02/cir11402.htm [ Links ]
22. Mittendorf EA, Evans DB, Lee JE, Perrier ND. Pheochromocytoma: Advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am. 2007;21:509-25. [ Links ]
23. Tamm EP, Kim EE, Ng CS. Imaging of neuroendocrine tumors. Hematol Oncol Clin North Am. 2007;21:409-32. [ Links ]
24. Janetschek G, Finkenstedt G, Gasser R, Waibel UG, Peschel R, Bartsch G, et al. Laparoscopic surgery for pheochromocytoma: Adrenalectomy, partial resection, excision of paragangliomas. J Urol. 1998;160:330-4. [ Links ]
25. Cascon A, Pita G, Burnichon N, Landa I, López-Jiménez E, Montero-Conde C, et al. Genetics of pheochromocytoma and paraganglioma in Spanish patients. J Clin Endocrinol Metab. 2009;94:1701-5. [ Links ]
26. Timmers HJ, Kozupa A, Eisenhofer G, Raygada M, Adams KT, Solis D, et al. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2007;92:779-86. [ Links ]
27. Malone MJ, Libertino JA, Tsapatsaris NP, Woods BO. Preoperative and surgical management of pheochromocytoma. Urol Clin North Am. 1989;16:567-82. [ Links ]
28. Ilias I, Pacak K. A clinical overview of pheochromocytomas/paragangliomas and carcinoid tumors. Nucl Med Biol. 2008;35(Suppl.1):S27-34. [ Links ]
29. Yu R, Nissen NN, Chopra P, Dhall D, Phillips E, Wei M. Diagnosis and treatment of pheochromocytoma in an academic hospital from 1997 to 2007. Am J Med. 2009;122:85-95. [ Links ]
30. Brain KL, Kay J, Shine B. Measurement of urinary metanephrines to screen for pheochromocytoma in an unselected hospital referral population. Clin Chem. 2006;52:2060-4. [ Links ]
31. Goldstein DS, Eisenhofer G, Flynn JA, Wand G, Pacak K. Diagnosis and localization of pheochromocytoma. Hypertension. 2004;43:907-10. [ Links ]
32. Kimura N, Watanabe T, Noshiro T, Shizawa S, Miura Y. Histological grading of adrenal and extra-adrenal pheochromocytomas and relationship to prognosis: A clinicopathological analysis of 116 adrenal pheochromocytomas and 30 extra-adrenal sympathetic paragangliomas including 38 malignant tumors. Endocr Pathol. 2005;16:23-32. [ Links ]
33. Fernández-Cruz L, Taura P, Saenz A, Benarroch G, Sabater L. Laparoscopic approach to pheochromocytoma: Hemodynamic changes and catecholamine secretion. World J Surg. 1996;20:762-8. [ Links ]
34. Quint LE, Glazer GM, Francis IR, Shapiro B, Chenevert TL. Pheochromocytoma and paraganglioma: Comparison of MR imaging with CT and I-131 MIBG scintigraphy. Radiology. 1987;165:89-93. [ Links ]
35. Havekes B, Lai EW, Corssmit EP, Romijn JA, Timmers HJ, Pacak K. Detection and treatment of pheochromocytomas and paragangliomas: Current standing of MIBG scintigraphy and future role of PET imaging. Q J Nucl Med Mol Imaging. 2008;52:419-29. [ Links ]
36. van der Harst E, de Herder W, Bruining H, Jaap Bonjer H, de Krijger R, Lamberts S. [123I]Metaiodobenzylguanidine and [111In]Octreotide uptake in benign and malignant pheochromocytomas. J Clin Endocrinol Metab. 2001;86:685-93. [ Links ]
37. van Der Horst-Schrivers AN, Jager Ol, Boenzen HM, Schouten JP, Kema IP, Links TP. Iodine-123 metaiodobenzylguanidine scintigraphy in localising phaeochromocytomas-experience and meta-analysis. Anticancer Research, 2006;26:1599-604. [ Links ]
38. Kercher KW, Novitsky YW, Park A, Matthews BD, Litwin DE, Heniford BT. Laparoscopic curative resection of pheochromocytomas. Ann Surg. 2005;241:919-26. [ Links ]
39. Tiyadath BN, Sukumar S, Saheed CS, Hattangadi SB. Laparoscopic adrenalectomy - is it any different in phaeochromocytoma and non-phaeochromocytoma? Asian J Surg. 2007;30:244-9. [ Links ]
40. Misra MC, Bhattacharjee HK, Hemal AK, Bansal VK. Laparoscopic management of rare retroperitoneal tumors. Surg Laparosc Endosc Percutan Tech. 2010;20:e117-22. [ Links ]
41. Hwang J, Shoaf G, Uchio EM, Watson J, Pacak K, Linehan WM, et al. Laparoscopic management of extra-adrenal pheochromocytoma. J Urol. 2004;171:72-6. [ Links ]
42. Walz MK, Peitgen K, Neumann HP, Janssen OE, Philipp T, Mann K. Endoscopic treatment of solitary, bilateral, multiple, and recurrent pheochromocytomas and paragangliomas. World J Surg. 2002;26:1005-12. [ Links ]
43. Edwin B, Kazaryan AM, Mala T, Pfeffer PF, Tonnessen TI, Fosse E. Laparoscopic and open surgery for pheochromocytoma. BMC Surg. 2001;1:2. [ Links ]
44. Mellon MJ, Sethi A, Sundaram CP. Laparoscopic adrenalectomy: Surgical techniques. Indian J Urol. 2008;24:583-9. [ Links ]
45. Kazaryan AM, Kuznetsov NS, Shulutko AM, Beltsevich DG, Edwin B. Evaluation of endoscopic and traditional open approaches to pheochromocytoma. Surg Endosc. 2004;18:937-41. [ Links ]
46. Eisenhofer G, Bornstein SR, Brouwers FM, Cheung NK, Dahia PL, de Krijger RR, et al. Malignant pheochromocytoma: Current status and initiatives for future progress. Endocr Relat Cancer. 2004;11:423-36. [ Links ]
47. Scholz T, Eisenhofer G, Pacak K, Dralle H, Lehnert H. Clinical review: Current treatment of malignant pheochromocytoma. J Clin Endocrinol Metab. 2007;92:1217-25. [ Links ]
48. Kumaki N, Kajiwara H, Kameyama K, DeLellis RA, Asa SL, Osamura RY, et al. Prediction of malignant behavior of pheochromocytomas and paragangliomas using immunohistochemical techniques. Endocr Pathol. 2002;13:149-56. [ Links ]
49. Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score PASS) to separate benign from malignant neoplasms: A clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551-66. [ Links ]
50. Bedard E, Bergeron S, Poirier P, Dumesnil JG. Pheochromocytoma associated with apical-sparing left ventricular dysfunction due to acute afterload mismatch: A novel clinical entity? Can J Cardiol. 2007;23:1157-8. [ Links ]
51. Bernini G, Galetta F, Franzoni F, Bardini M, Taurino C, Moretti A, et al. Normalization of catecholamine production following resection of phaeochromocytoma positively influences carotid vascular remodelling. Eur J Endocrinol. 2008;159:137-43. [ Links ]
52. Kim HS, Chang WI, Kim YC, Yi SY, Kil JS, Hahn JY, et al. Catecholamine cardiomyopathy associated with paraganglioma rescued by percutaneous cardiopulmonary support: Inverted Takotsubo contractile pattern. Circ J. 2007;71:1993-5. [ Links ]
53. López M, Águila P, González E. Anestesia en el feocromocitoma: presentación de un paciente. Revista Cubana de Anestesiología y Reanimación. 2004;2:45-8. [ Links ]
54. Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92:4069-79. [ Links ]
55. Sesay M, Tauzin-Fin P, Gosse P, Ballanger P, Maurette P. Real-time heart rate variability and its correlation with plasma catecholamines during laparoscopic adrenal pheochromocytoma surgery. Anesth Analg. 2008;106:164-70. [ Links ]
56. Carvalho MR, Dias T, Rodrigues A, Machado AP, Esteves R, do C, I. Alpha blockade with doxazosin in pheochromocytoma –report of three cases. Rev Port Cardiol. 2010;29:299-308. [ Links ]
57. Chiu AW. Laparoscopic retroperitoneal adrenalectomy: Clinical experience with 120 consecutive cases. Asian J Surg. 2003;26:139-44. [ Links ]
58. Rabii R, Salomon L, Saint F, Hoznek A, Cicco A, Chopin D, et al. Treatment of pheochromocytomas with retroperitoneal laparoscopy. Prog Urol. 2001;11:16-20. [ Links ]
59. Humphrey R, Gray D, Pautler S, Davies W. Laparoscopic compared with open adrenalectomy for resection of pheochromocytoma: A review of 47 cases. Can J Surg. 2008;51:276-80. [ Links ]
60. Kasahara T, Nishiyama T, Takahashi K. Laparoscopic adrenalectomy for pheochromocytoma: Evaluation of experience and strategy at a single institute. BJU Int. 2009;103:218-22. [ Links ]
61. Tauzin-Fin P, Sesay M, Gosse P, Ballanger P. Effects of perioperative alpha1 block on haemodynamic control during laparoscopic surgery for phaeochromocytoma. Br J Anaesth. 2004;92:512-7. [ Links ]
62. O’Riordain DS, Young WF Jr, Grant CS, Carney JA, van Heerden JA. Clinical spectrum and outcome of functional extraadrenal paraganglioma. World J Surg. 1996;20:916-21. [ Links ]
63. Shirkare S, Kuwert T, Weckesser M, Czech N, Langen KJ, Muller-Gartner HW. Localization of a pheochromocytoma using I-123 MIBG adrenal scintigraphy. J Postgrad Med. 1994;40:85-7. [ Links ]
64. Chen L, Li F, Zhuang H, Jing H, Du Y, Zeng Z. 99mTc-HYNIC-TOC scintigraphy is superior to 131I-MIBG imaging in the evaluation of extraadrenal pheochromocytoma. J Nucl Med. 2009;50:397-400. [ Links ]
65. Suh IW, Lee CW, Kim YH, Hong MK, Lee JW, Kim JJ, et al. Catastrophic catecholamine-induced cardiomyopathy mimicking acute myocardial infarction, rescued by extracorporeal membrane oxygenation ECMO) in pheochromocytoma. J Korean Med Sci. 2008;23:350-4. [ Links ]
66. Neumann HP, Eng C. The approach to the patient with paraganglioma. J Clin Endocrinol Metab. 2009;94:2677-83. [ Links ]
67. Benn DE, Robinson BG. Genetic basis of phaeochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab. 2006;20:435-50. [ Links ]
68. Jimenez C, Cote G, Arnold A, Gagel RF. Review: Should patients with apparently sporadic pheochromocytomas or paragangliomas be screened for hereditary syndromes? J Clin Endocrinol Metab. 2006;91:2851-8. [ Links ]
69. Boedeker CC, Neumann HP, Offergeld C, Maier W, Falcioni M, Berlis A, et al. Clinical features of paraganglioma syndromes. Skull Base. 2009;19:17-25. [ Links ]
70. Bauters C, Vantyghem MC, Leteurtre E, Odou MF, Mouton C, Porchet N, et al. Hereditary phaeochromocytomas and paragangliomas: A study of five susceptibility genes. J Med Genet. 2003;40:e75. [ Links ]
71. Asari R, Scheuba C, Kaczirek K, Niederle B. Estimated risk of pheochromocytoma recurrence after adrenal-sparing surgery in patients with multiple endocrine neoplasia type 2A. Arch Surg. 2006;141:1199-205. [ Links ]
72. Amar L, Servais A, Gimenez-Roqueplo AP, Zinzindohoue F, Chatellier G, Plouin PF. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab. 2005;90:2110-6. [ Links ]

