










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cirugía
Print version ISSN 2011-7582On-line version ISSN 2619-6107
rev. colomb. cir. vol.26 no.2 Bogotá Apr./June 2011
(1) Estudiante de Medicina, X semestre, Universidad de los Andes, Bogotá, D.C., Colombia.
(2) Jefe, Servicio de Trasplante Hepático y Cirugía Biliopancreática, Hospital Universitario Fundación Santa Fe de Bogotá, Bogotá, D.C., Colombia.
(3) Jefe, Servicio de Endocrinología, Hospital Universitario Fundación Santa Fe de Bogotá, Bogotá, D.C., Colombia.
(4) Departamento de Patología, Hospital Universitario Fundación Santa Fe de Bogotá, Bogotá, D.C., Colombia.
(5) Grupo de Genética y Oncología Molecular, Instituto Nacional de Cancerología, Bogotá, D.C., Colombia.
(6) Servicio de Endocrinología, Hospital Universitario Fundación Santa Fe de Bogotá, Bogotá, D.C., Colombia.
Correspondencia: Lucía Beatriz Taboada, Bogotá, Colombia. Correo electrónico: lb.taboada57@uniandes.edu.co
Fecha de recibido: 28 de febrero de 2011. Fecha de aprobación: 28 de marzo de 2011.
Resumen
La menina es una proteína supresora de tumor codificada por el gen MEN1, cuya mutación produce procesos neoplásicos en múltiples tejidos del organismo que pueden manifestarse por generaciones como síndromes familiares. La mutación genera un espectro de enfermedad que va desde el hiperparatiroidismo familiar aislado hasta la neoplasia endocrina múltiple de tipo 1, caracterizada por neoplasias de paratiroides, hipófisis anterior, páncreas endocrino y duodeno, entre otras.
Como ejemplo, se presentan dos casos de pacientes con neoplasias endocrinas secundarias a la mutación del gen MEN1. Se revisa la información actual sobre la etiopatogenia y carcinogénesis entendidos apenas recientemente, al igual que otras mutaciones involucradas en los síndromes neoplásicos expuestos y se dan unas recomendaciones finales.
Palabras clave: menina; neoplasia endocrina múltiple; hiperparatiroidismo primario; prolactinoma; carcinoma neuroendocrino.
Abstract
Menin is a tumor suppressor protein, encoded by the MEN1 gene, whose mutation can generate neoplastic disease in multiple tissues of the human body, which for generations can manifest as familial syndromes. The mutation generates a spectrum of diseases ranging from familial isolated hyperparathyroidism to multiple endocrine neoplasia type 1, characterized by neoplasm of parathyroid glands, anterior pituitary, endocrine pancreas and duodenum, among others.
We present two cases of patients with endocrine neoplastic disease secondary to menin’s mutation. We review current information regarding its ethiopathogeny and its mechanism of carcinogenesis just recently understood. Additionally we review other mutations involved in the neoplastic syndromes exposed and present some final recommendations.
Key words: menin; multiple endocrine neoplasia; hyperparathyroidism, primary; prolactinoma; carcinoma, neuroendocrine.
Introducción
Los síndromes neoplásicos hereditarios son responsables del 5 al 10% de todos los cánceres, aproximadamente (1). En la mayoría de los casos se producen por una mutación que inactiva un gen de supresión tumoral y pocos casos se deben a mutaciones que activan oncogenes. Entre los síndromes neoplásicos hereditarios, se hallan los causados por la mutación del gen MEN1 que codifica para la menina, una proteína supresora de tumores. En la mayoría de los casos, su mutación lleva a la aparición de la neoplasia endocrina múltiple de tipo 1; sin embargo, se han encontrado mutaciones en el gen MEN1 en pacientes con hiperparatiroidismo familiar aislado (2).
La neoplasia endocrina múltiple de tipo 1 es un síndrome genético, autosómico dominante con una alta penetrancia, estimada en 80% a los 50 años de edad; la mayoría de los pacientes se diagnostican al final de la juventud (3). Se caracteriza principalmente por neoplasias de glándula paratiroides, hipófisis anterior, páncreas endocrino y duodeno. También, puede asociarse con tumores neuroendocrinos de pulmón, tubo digestivo y timo, tumores de corteza suprarrenal, lipomas múltiples y angiofibromas (4).
La presentación clínica de esta enfermedad es muy diversa y depende en gran parte del tejido endocrino afectado; la condición más comúnmente expresada es el hiperparatiroidismo primario, que está presente en alrededor de 95% de los pacientes (5), mientras que el porcentaje de pacientes con neoplasia endocrina múltiple de tipo 1 en pacientes con hiperparatiroidismo primario, es de 1 a 5% (6).
Para el diagnóstico de la neoplasia endocrina múltiple de tipo 1, se requiere la presencia de tumor en dos de los tres principales órganos endocrinos relacionados (paratiroides, tejido endocrino enteropancreático, pituitaria anterior) y se considera un síndrome familiar si hay, al menos, un caso de pariente en primer grado con mínimo uno de los tumores principales (7).
Por su parte, el hiperparatiroidismo familiar aislado se define como hiperparatiroidismo hereditario sin otras neoplasias o endocrinopatías asociadas; su forma de herencia es autosómica dominante y se caracteriza por tumores paratiroideos benignos únicos o multiglandulares (8). Se presenta usualmente en una edad más temprana y corresponde sólo a 1% de los casos de hiperparatiroidismo (9).
Las bases genéticas de esta enfermedad están menos claras. Las mutaciones en diversos genes pueden llevar a esta condición, entre ellas, la mutación de MEN1, aunque en muchos casos no se ha logrado identificar el gen causante.
Nuestro objetivo fue revisar la información actual relacionada con el mecanismo de carcinogénesis involucrado en la mutación del MEN1, la etiopatogenia del hiperparatiroidismo familiar aislado asociada a este trastorno y otras alteraciones genéticas presentes en la neoplasia endocrina múltiple de tipo 1, y presentar como ejemplo dos casos clínicos de tumores endocrinos originados por mutaciones en el gen MEN1, el primero correspondiente a hiperparatiroidismo familiar aislado y el segundo, a neoplasia endocrina múltiple de tipo 1.
Descripción de los casos
Caso 1: hiperparatiroidismo familiar aislado
Se trata de una mujer de 54 años con antecedentes de hipotiroidismo primario e hiperparatiroidismo primario recidivante, diagnosticado en el año 2000, que requirió paratiroidectomía en 2001 y confirmó un adenoma de la glándula paratiroides inferior izquierda; en 2002, por persistencia de hipercalcemia, requirió otra intervención que demostró un nuevo adenoma.
En 2003, ingresó para control y se encontraron niveles de parathormona (PTH) de 91,5 pg/ml (valor normal, 15 a 68 pg/ml), de calcio ionizado de 1,29 mmol/L (valor normal, 1,16 a 1,32 mmol), de TSH de 18,3 mUI/ml (valor normal, 0,4 a 4,0 mlU/L) y densitometría ósea normal, medida por DXA (Dual-energy X-ray Absorptiometry).
Se inició tratamiento con carbonato de calcio, 1.500 mg dos veces al día, hasta junio de 2008, cuando se consideró que la paciente cursaba nuevamente con hiperparatiroidismo activo leve debido a niveles elevados de paratohormona (91 pg/ml), hipercalcemia leve (10,4 mg/dl) y densitometría ósea con discreta disminución de la masa ósea al compararla con las previas.
En la gammagrafía de paratiroides se encontró un adenoma paratiroideo por debajo del polo inferior del lóbulo izquierdo de la tiroides.
Fue llevada a junta médica en julio de 2008, en la que determinaron no practicar intervención quirúrgica en el momento, debido a que la enfermedad no tenía gran relevancia clínica, y se dejó en seguimiento. Se completaron los estudios que mostraron niveles normales de 25 OH vitamina D en 36,3 ng/ml y se investigó la posibilidad de una neoplasia endocrina múltiple de tipo 1, la que se descartó mediante resonancia magnética (RM), en la que se observó una hipófisis normal, y los niveles de prolactina y somatomedina C fueron normales. La tomografía computadorizada (TC) de abdomen fue normal y no había manifestaciones clínicas de aumento de la producción de hormonas pancreáticas (hipoglucemia, hipergastrinemia, entre otros).
En marzo de 2010, a un sobrino de la paciente, con hipercalcemia por hiperparatiroidismo primario, se le confirmó la mutación en el gen MEN1. Posteriormente, se confirmó la misma mutación en la paciente y su hermana. En el estudio de sangre periférica se identificó la variante familiar MEN1:c.665A>C en el exón 4 del gen MEN1 en estado heterocigoto mediante GENDIA Genetic Diagnostic Network.
Esta substitución es una variación “sin sentido”; se predice que conduce a la substitución de una tirosina por una serina en la posición del aminoácido 222 de la proteína resultante MEN 1 (MEN1: pTYR222Ser).
La variante MEN1: c.665A>C es nueva y no ha sido descrita previamente en otros pacientes ni controles. Por esta razón, su significancia clínica no es aún clara.
Con este hallazgo, se confirmó la presencia de hiperparatiroidismo familiar y se advirtió la posibilidad de que, potencialmente en el tiempo y de manera asincrónica, puedan aparecer otro tipo de neoplasias de hipófisis anterior y de páncreas, por lo cual es necesario el control anual permanente de las personas afectadas y de los no afectados cada tres años. Actualmente, la enfermedad la presentan ocho hermanos, la madre y un sobrino de la paciente (genograma 1), y no se han demostrado hasta el momento otras neoplasias endocrinas en esta familia.
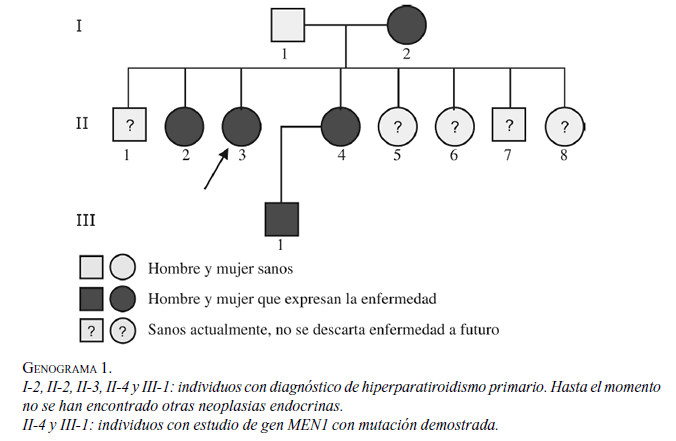
La paciente cursa con niveles normales de calcio ionizado y paratohormona persistentemente elevada pero estable. En los últimos exámenes de laboratorio, tomados en abril de 2010, se encontró calcio ionizado de 1,32 mmol/L, PTH de 128 pg/ml, gastrina de 15,7 pg/ml (normal); RM de silla turca, normal; en la TC de abdomen y pelvis se observaron quistes hepáticos simples, sin lesiones sospechosas de enfermedad de páncreas ni de las suprarrenales.
Caso 2: neoplasia endocrina múltiple de tipo 1
Se trata de un hombre de 48 años con historia de carcinoma neuroendocrino de páncreas, metastásico a hígado, diagnosticado en 2007 y tratado con múltiples esquemas de quimioterapia, sin buenos resultados, que ingresó en septiembre de 2009 para nuevas propuestas terapéuticas.
Entre los antecedentes, presentaba gastritis crónica de varios años de evolución, hipotiroidismo primario en tratamiento con suplemento, hipertensión arterial en tratamiento con losartán e historia de urolitiasis con litotripsia renal izquierda. En el 2008, se le resecó un lipoma en la región abdominal. En ese mismo año, se visualizó en la TC de tórax un nódulo pulmonar izquierdo de 15 mm, con características benignas.
Entre los antecedentes familiares, el padre murió a los 40 años por hemorragia de vías digestivas altas, la madre de 70 años estaba viva, con hipertensión arterial sistémica y cardiopatía. De los seis hermanos, uno murió durante el posoperatorio por una úlcera péptica perforada a los 24 años, una hermana murió por cáncer genitourinario metastásico a columna a los 50 años, y en el momento, una hermana de 37 años presentaba urolitiasis e hiperpartiroidismo primario en estudio por posible neoplasia endocrina múltiple de tipo 1. Tenía un hijo sano en el momento (genograma 2).
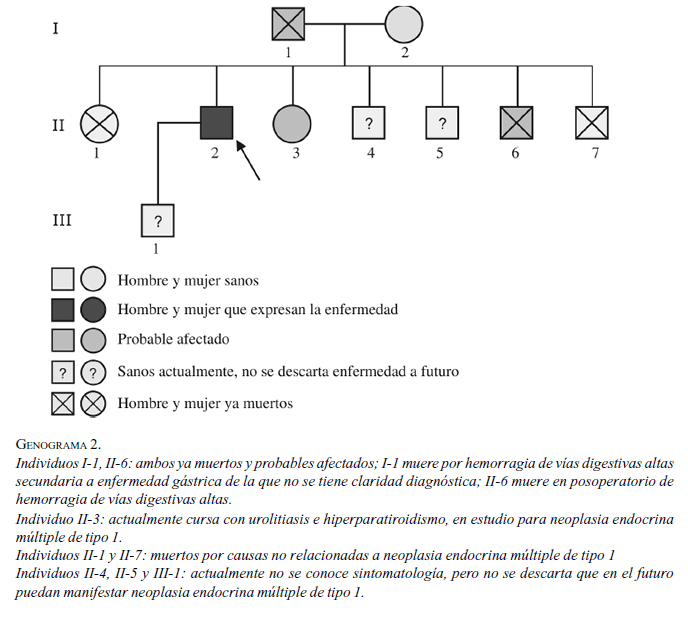
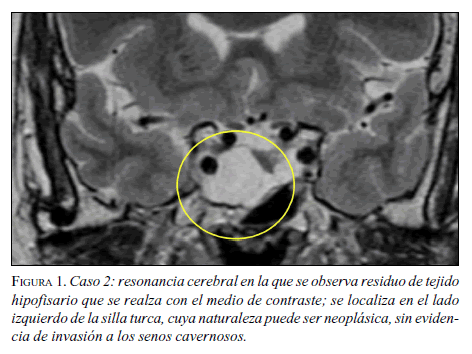
Con la historia clínica descrita, se sospechó una neoplasia endocrina múltiple de tipo 1, se caracterizó desde el punto de vista de las hormonas y se hicieron los estudios radiológicos correspondientes. Las imágenes de la RM cerebral revelaron un macroadenoma hipofisiario y el panel de hormonas hipofisiarias mostró elevación de prolactina, sin compromiso de las otras líneas hormonales (figura 1). El valor máximo de prolactina fue de 350 ng/ml, para lo cual se inició cabergolina hasta requerir 0,5 mg dos veces semanales para control hormonal.
La densitometría ósea DXA demostró deterioro de la masa ósea asociado a urolitiasis por hiperparatiroidismo primario, con niveles elevados de PTH y calcio ionizado (172,80 pg/ml y 1,67 mmol/L, respectivamente), y nivel normal de 25 OH vitamina D en 33,5 ng/ml (figuras 2A y 2B); se inició corrección de la hipercalcemia con hidratación y bisfosfonatos orales. La gammagrafía con 99mTecnecio-sestamibi de paratiroides mostró adenomas paratiroideos, uno localizado por debajo del polo inferior del lóbulo izquierdo de la tiroides y el otro en el lado derecho (figura 2C).
Se resecaron cuatro glándulas paratiroides y se reimplantó una de ellas en el cuello, en febrero de 2010, con hallazgos patológicos correspondientes a cambios por hiperplasia. Se descartó neoplasia paratiroidea y la coloración de rojo Sudán confirmó el diagnóstico (figura 2D).
En la TC de abdomen y la gammagrafía con octreótido se observó una masa pancreática metastásica a hígado (figuras 3A y 3B). En la biopsia hepática se reportó un carcinoma neuroendocrino metastásico, con reacción intensa de células tumorales para la somatostatina y la sinaptofisina y CAM 5.2 (figura 3C), ocasionales células tumorales reactivas para cromogranina, con resultados negativos para serotonina, glucagón, CDX2, insulina, factor VII y CD34, e índice de proliferación celular por Ki 67 de 5%.
En julio de 2010, el paciente fue intervenido para metastasectomía hepática y pancreatectomía distal más esplenectomía. El estudio de histopatología reportó un carcinoma neuroendocrino bien diferenciado, con tamaño tumoral de 5,3 x 4,5 x 4,0 cm y reacción de las células tumorales para cromogranina, sinaptofisina, CAM5.2, CK19 difusa e intensa, Ki-67 del 20%, y conteo mitótico 3 por 10 campos de gran aumento, negativo para glucagón, somatostatina e insulina. En el parénquima pancreático no tumoral se encontraron múltiples microadenomas de las células de Langerhans, que medían entre 0,4 y 0,5 cm, y que igualmente presentaban reacción con la cromogranina y la sinaptofisina.
Mediante análisis de las mutaciones por secuencia directa de la región codificante del MEN1 en sangre periférica, se encontró una mutación deletérea c619 del ACAG en el exón 3, la cual conduce a la pérdida de cuatro pares de bases (ACAG) correspondientes al codón 210 (ACA) y a la primera base del codón 211 (GTC), generando un codón de parada 13 tripletas delante de la deleción. Actualmente, se tiene controlada la hipersecreción hormonal y no hay enfermedad en progresión (tabla 1).
Revisión del tema
Mutación del gen de la menina
La menina es una proteína nuclear reguladora del ciclo celular que juega un papel importante en las vías de diferenciación y crecimiento celular (10). Es codificada por el gen MEN1 ubicado en el cromosoma 11q13 y es ampliamente expresada tanto en tejido endocrino como no endocrino (11). La menina está predominantemente localizada en el núcleo celular, gracias a señales de localización nuclear que no sólo se encargan de llevarla hacia el núcleo sino que participan en la unión al ADN y en la coordinación de la expresión de ciertos genes (12).
En los últimos años se han hecho avances en el entendimiento de sus funciones en la regulación del ciclo celular y su papel como proteína supresora de tumores. La mutación inactiva su función como reguladora de la proliferación celular, lo que da lugar a la formación de tumores, algunos de ellos malignos (13).
Se han identificado mutaciones del gen MEN1 en familias con síndromes de cáncer hereditario, y el ejemplo más claro es la neoplasia endocrina múltiple de tipo 1, con más de 1.300 mutaciones reportadas (14). Esta alteración genética se ha visto asociada a un espectro de fenotipos expresados que varían desde casos que presentan únicamente hiperparatiroidismo familiar aislado, hasta los casos de neoplasia endocrina múltiple y otros síndromes con presentación más grave (15). Este espectro de manifestación de la mutación se puede evidenciar con los dos casos expuestos anteriormente.
A pesar de que no se ha logrado una correlación genotipo-fenotipo, se ha visto que en familias con hiperparatiroidismo aislado predominan las mutaciones de sentido errado, en las que un codón pasa a codificar para otro aminoácido, mientras que, en la neoplasia endocrina múltiple de tipo 1, predominan las mutaciones sin sentido en la que la mutación lleva a la aparición de un codón de parada (8, 16). Más de 70% de las mutaciones son sin sentido y generan una proteína truncada al impedir la transcripción del terminal C, alterando así las señales de localización nuclear y las funciones dependientes de éstas (12, 14).
Los tumores secundarios a mutaciones del gen MEN1 surgen como consecuencia del mecanismo de “dos eventos” (two-hit), siendo el primer evento una mutación germinal, ya sea heredada o de novo, y el segundo, una pérdida somática del otro alelo normal (1, 17); es decir, se requiere la inactivación bialélica del gen para el desarrollo de una célula tumoral (18). Esto contribuye a que la enfermedad tenga una expresión fenotípica variable entre los individuos, incluso entre gemelos idénticos (19). Esta pérdida de la heterocigocidad ocurre en la mayoría de síndromes de cáncer hereditario, especialmente en los secundarios a mutaciones de genes de supresión tumoral que necesitan la inactivación de las dos copias del gen para causar la enfermedad (1).
La mutación del gen MEN1 no es exclusiva de los síndromes hereditarios; también se ha visto implicada en ciertos tumores esporádicos, entre ellos tumores neuroendocrinos pancreáticos o bronquiales que frecuentemente la presentan, lo cual reafirma la importancia de la inactivación del MEN1 en los procesos de génesis de tumores (10). El 65% de los tumores carcinoides de timo muestran pérdida de la heterocigocidad en el gen, al igual que 48% de los carcinoides esporádicos de estómago y 36% de los carcinoides de pulmón (20).
La inactivación de la menina induce carcinogénesis por diversos mecanismos entendidos apenas recientemente. Es conocido su papel en la regulación de la transcripción, la proliferación celular, la apoptosis y el mantenimiento de la estabilidad del ADN.
Se ha demostrado que la menina tiene interacción con un gran número de factores de transcripción, entre ellos JunD y NF-kB, que llevan a la inhibición de la transcripción mediada por estas vías (11). Además, participa en la regulación de genes, como l IGFBP- 2 (Insulin-like Growth Factor Binding Protein 2), involucrado en la regulación de la proliferación celular.
En el ciclo celular, la menina cumple un papel regulador por medio de varios mecanismos, entre ellos, la inhibición de la expresión de la ciclina D1 y la D3, que forman una cinasa activa con la cinasa dependiente de ciclina 4 (CDK4) que promueve la progresión de G1 a S (21). Además, estimula la expresión de p18ink4c y p27kip1, que inhiben la actividad de la cinasa dependiente de la ciclina 2 (CDK2), lo que impide la transición desde la fase G0/1 a S. La pérdida de la menina altera la expresión de estos inhibidores y causa un crecimiento celular sin regulación (22, 23).
Asimismo, está implicada en la regulación de la apoptosis, induciendo de forma normal la expresión de caspasa 8, un componente en las vías de apoptosis. La inactivación del gen disminuye la expresión de caspasa 8 y aumenta la resistencia a la apoptosis inducida por el factor de necrosis tumoral alfa (TNFa) (24).
Por último, participa en el mantenimiento de la integridad del ADN y la estabilidad cromosómica en células bajo estrés, interactuando con proteínas involucradas en el mantenimiento de la estabilidad del genoma, como la histona metiltransferasa y la histona deacetilasa, modulando la actividad de proteínas involucradas con la replicación y reparación del ADN, tales como la Cdc7/ASK, y mediando la transcripción de genes relacionados con la reparación del ADN (21, 23).
Mutaciones relacionadas con hiperparatiroidismo familiar aislado
El hiperparatiroidismo primario es una endocrinopatía frecuente, con una incidencia estimada de 3 por 1.000 adultos (25). En la mayoría de los casos, aparece de forma esporádica y tiene como causa un adenoma paratiroideo único (80 a 85%) (26). Aproximadamente, 5% de los casos están asociados con síndromes de cáncer hereditario (25), que dan lugar al hiperparatiroidismo familiar primario. Esta condición es, generalmente, heredada de forma autosómica dominante y, a diferencia del adenoma común en los casos esporádicos, usualmente se manifiesta con hiperplasia multiglandular de paratiroides y a una edad más temprana (25, 27). Su presentación varía dentro de un gran espectro que va desde el hiperparatiroidismo familiar aislado hasta formas en las que el hiperparatiroidismo hace parte de síndromes que incluyen tumores de otros tejidos, como la neoplasia endocrina múltiple de tipo 1 y, menos frecuentemente, la de tipo 2A, la hipercalcemia hipocalciúrica familiar y el síndrome de hiperparatiroidismo familiar-tumor mandibular (15, 28).
El hiperparatiroidismo familiar aislado es una forma de hiperparatiroidismo que ocurre en ausencia de otras endocrinopatías, al menos, con un familiar en primer grado con hiperparatiroidismo confirmado y sin historia personal o familiar de neoplasia endocrina múltiple (29). Es una condición genéticamente heterogénea (16), en la que se han encontrado mutaciones en los genes MEN1, CASR y HRPT2, que normalmente dan lugar a los síndromes antes mencionados y que, en ciertos casos, pueden expresar como único fenotipo el hiperparatiroidismo familiar aislado (15), por lo que se considera un diagnóstico de exclusión una vez que se ha descartado cualquiera de los síndromes causantes de hiperparatiroidismo primario (30).
El hiperparatiroidismo familiar aislado se ha descrito en ocasiones como una expresión incompleta de síndromes como la neoplasia endocrina múltiple de tipo 1 y el síndrome de hiperparatiroidismo familiar-tumor mandibular que cursan usualmente con hiperparatiroidismo primario (31, 32). Sin embargo, en la mayoría de los pacientes con hiperparatiroidismo familiar aislado no se encuentran mutaciones germinales en los genes MEN1, HRPT2 o CASR (33), por lo que muchos autores lo consideran una entidad separada y clínicamente distinta (29). Esto deja la posibilidad de nuevos genes involucrados aún por identificar.
El gen PRAD1 es un protooncogén perteneciente a la familia de las ciclinas; su mutación induce la expresión aumentada de ciclina D1, lo que promueve la progresión del ciclo celular. Se han reportado mutaciones de este oncogén en casos de adenomas esporádicos, pero no se han encontrado mutaciones cromosómicas germinales que involucren este gen en las formas familiares de hiperparatiroidismo primario (33, 34).
A pesar de que no se ha logrado aislar un gen, se ha propuesto una región en el cromosoma 2. En el análisis de 10 árboles genealógicos de familias con hiperparatiroidismo familiar aislado y de 59 individuos con genotipificación con hiperparatiroidismo familiar aislado después de excluir mutaciones en MEN1, CASR y HRTP2, se precisó que el gen causante podría estar en una región de 1.7 Mb en el cromosoma 2p13.3-14 (9).
Se ha detectado pérdida cromosómica de ADN en 1p, 6q, 9p y 13q, en tumores paratiroideos tanto benignos como malignos, lo que indica la presencia potencial de genes supresores de tumor en estas regiones (33).
Por el momento, se continúa en la búsqueda de otras alteraciones genéticas que sean responsables del hiperparatiroidismo familiar aislado.
Otras mutaciones responsables de la neoplasia endocrina múltiple de tipo 1
De 20 a 25% de los pacientes con neoplasia endocrina múltiple de tipo 1 no poseen mutaciones en el gen MEN1 (35). Esto ha abierto la posibilidad de que mutaciones germinales en otros genes sean responsables del síndrome, y de que otros factores interactúen con la menina para lograr completamente su función de supresora tumoral (36).
Los estudios han apuntado a dar como posible causa mutaciones en ciertos genes reguladores del ciclo celular, especialmente en los genes inhibitorios de las cinasas dependientes de ciclina (Cyclin-Dependent Kinase Inhibitors, CDKI). Éstas son fundamentales para la proliferación celular, al promover la entrada de la célula a la fase G1 y la transición G1 a S (37). Entre los inhibidores de estas cinasas, encontramos la familia INK4, constituida por p16, p15, p18, p19 (p16Ink4a, p15Ink4b, p18Ink4c, y p19Ink4d), y la familia Cip/Kip, constituida por p21, p27 y p57 (p21Cip1, p27Kip1, y p57Kip2) (37).
Los ratones con mutaciones germinales en CDKN1B (p27) tienen predisposición al desarrollo de tumores endocrinos y fenotípicamente expresan características de la neoplasia endocrina múltiple de tipos 1 y 2 (38). En humanos, se han reportado pocos casos de mutaciones germinales en el gen CDKN1B en pacientes con fenotipo de la neoplasia endocrina múltiple de tipo 1 (35, 39). Marinoni et al. reportaron seis casos de mutaciones en el gen CDKN1B en pacientes con fenotipos de pseudoneoplasia endocrina múltiple, y negativos para mutaciones en MEN1 o RET (40). Aunque la presencia de esta mutación es rara, se ha considerado como causante de una nueva forma de neoplasia endocrina múltiple llamada neoplasia endocrina múltiple de tipo 4, cuyas características clínicas aún están por definirse debido al pequeño número de casos (41).
Agarwal et al. seleccionaron 196 casos de neoplasia endocrina múltiple de tipo 1 en los que no se hubiese identificado mutación en MEN1, y analizaron cada uno de los siete genes para CDKI. Obtuvieron como resultado 7 probables mutaciones en 4 de los genes (p15, p18, p21 y p27), que podrían ser causa de la enfermedad y otros síndromes relacionados; sin embargo, las frecuencias de estas mutaciones siguen siendo muy bajas y oscilan entre 0,5 y 1,5%, y su importancia en la clínica está aún por definirse (37).
El gen de supresión tumoral AIP (Hydrocarbon receptor-Interacting Protein) ha sido recientemente identificado como un posible responsable de 15 a 30% de los casos de adenomas pituitarios familiares aislados; sin embargo, aunque se ha intentado detectar su mutación en pacientes con neoplasia endocrina múltiple de tipo 1, no hay casos reportados (38).
Estudio de mutaciones para menina 1
Se recomienda realizar un análisis de mutación para el gen MEN1 cuando se presente un sujeto con dos o más neoplasias endocrinas relacionadas con la neoplasia endocrina múltiple de tipo 1, cuando un sujeto haga parte de un grupo familiar afectado por dicha enfermedad, y cuando existan neoplasias endocrinas en dos o más miembros de la familia que hagan parte de su espectro, como el hiperparatiroidismo familiar.
Sin embargo, en la actualidad existe gran limitación de datos sobre el alcance real de la secuenciación de los genes implicados en el cáncer endocrino y no endocrino. Se desconoce lo siguiente:
- el número de sujetos con neoplasias endocrinas aparentemente esporádicas, que portan una mutación germinal potencialmente heredable; este fenómeno se ha reconocido ampliamente en el síndrome paraganglioma/feocromocitoma (42);
- la frecuencia con que se presenta una mutación germinal de novo;
- la sensibilidad necesaria para detectar una mutación en una población dada, la especificidad de las pruebas, y los falsos positivos y negativos de las diferentes técnicas de secuenciación (43);
- el porcentaje de distribución de las mutaciones responsables del hiperparatiroidismo familiar primario y de la neoplasia endocrina múltiple de tipo 1; por ejemplo, no se sabe cuántos sujetos podrían ser portadores de una mutación MEN1, CASR, HRTP2, y cuál estudio sería más costo-efectivo;
- cuáles variantes confieren mayor riesgo de aparición de neoplasias y cáncer en un sujeto o grupo familiar afectado, y la contribución de las variantes polimórficas a la expresión de la enfermedad en las formas sutiles, que podría ser el caso familiar 1;
- la contribución de la epigenética en la expresión de la enfermedad.
Recomendaciones
Los avances genéticos y la aplicación de la tecnología genética han modificado ostensiblemente el enfoque de las enfermedades oncológicas y no oncológicas, desde el punto de vista de prevención, promoción, diagnóstico, pronóstico, tratamiento y la misma investigación; estos dos casos son ilustrativos de cómo la alteración de un mismo gen puede generar diferentes enfermedades benignas y malignas, con abordajes médico-quirúrgicos simples o complejos, y seguimientos clínicos, hormonales y radiológicos convencionales o muy especializados.
Para poder establecer un diagnóstico adecuado en los síndromes heredofamiliares, es necesario realizar una historia clínica completa, en la que se incluyan con detalle los antecedentes familiares.
Se recomienda investigar la presencia de mutaciones del MEN1 en pacientes con hiperparatiroidismo a temprana edad, con hiperplasia de múltiples glándulas paratiroideas, con presencia de tumores relacionados con neoplasia endocrina múltiple de tipo 1, coexistentes o en el pasado o en un familiar (27).
Una vez se identifique la mutación genética, se debe brindar la asesoría genética correspondiente al sujeto portador; se debe buscar la mutación en los familiares en primer grado del caso índice. El análisis de ADN para MEN1 beneficia a todos los familiares enfermos y asintomáticos, incluso en la niñez (44).
Es necesario conocer el espectro clínico de esta enfermedad y tratar de hacer una correlación genotipo-fenotipo en la medida que sea posible, para plantear una propuesta diagnóstica, quirúrgica y de seguimiento a largo plazo.
La detección de mutaciones en el gen MEN1 en pacientes con hiperparatiroidismo primario aislado, es importante porque el tratamiento quirúrgico difiere en estos pacientes y, además, se deben investigar otros tumores relacionados con dicho gen.
En el análisis de las mutaciones del MEN1, un resultado negativo no descarta la enfermedad. En este caso, para los individuos con 50% de riesgo (con un familiar en primer grado con diagnóstico de neoplasia endocrina múltiple de tipo 1) se recomienda la evaluación clínica y bioquímica rutinarias, y una nueva tamización años más tarde, en la que se espera un análisis de nuevas mutaciones (44). En el caso puntual del hiperparatiroidismo, se puede hacer un análisis de otros genes involucrados en la enfermedad familiar.
Se requiere un trabajo multidisciplinario y de colaboración entre todas las especialidades clínicas y de investigación, para conocer el impacto en la población de la carga genética en estas enfermedades (45).
Debemos familiarizarnos con los diferentes estudios genéticos y, a la vez, promocionarlos y conocer sus alcances y sus debilidades en la práctica clínica.
Referencias1. Busygina V, Bale AE. Multiple endocrine neoplasia type 1 (MEN1) as a cancer predisposition syndrome: Clues into the mechanisms of MEN1-related carcinogenesis. Yale J Biol Med. 2006;79:105-14. [ Links ]
2. Cardinal JW, Bergman L, Hayward N, Sweet A, Warner J, Marks L, et al. A report of a national mutation testing service for the MEN1 gene: Clinical presentations and implications for mutation testing. J Med Genet. 2005;42:69-74. [ Links ]
3. Callender GG, Rich TA, Perrier ND. Multiple endocrine neoplasia syndromes. Surg Clin North Am. 2008;88:863-95. [ Links ]
4. Marini F, Falchetti A, Del Monte F, Carbonell Sala S, Gozzini A, Luzi E, et al. Multiple endocrine neoplasia type I. Orphanet J Rare Dis. 2006;1:38. [ Links ]
5. Wohllk N, Becker P, Véliz J, Pineda G. Neoplasias endocrinas múltiples: un modelo clínico para aplicar técnicas de genética molecular. Rev Méd Chile 2000;128:791-800. [ Links ]
6. Quevedo V, Soutelo M, Faraj G, Lutfi R. ¿NEM tipo 1 o una simple asociación entre dos adenomas hiperfuncionantes? Glánd Tir Paratir. 2009;18:28-32. [ Links ]
7. White ML, Doherty GM. Multiple endocrine neoplasia. Surg Oncol Clin N Am. 2008;17:439-59. [ Links ]
8. Villablanca A, Calender A, Forsberg L, Höög A, Cheng JD, Petillo D, et al. Germline and de novo mutations in the HRPT2 tumour suppressor gene in familial isolated hyperparathyroidism (FIHP). J Med Genet. 2004;41:e32. [ Links ]
9. Warner J, Nyholt D, Busfield F, Epstein M, Burgess J, Stranks S, et al. Familial isolated hyperparathyroidism is linked to a 1.7Mb region on chromosome 2p13.3-14. J Med Genet. 2006;43:e12. [ Links ]
10. Phan A, Yao J. Neuroenocrine tumors: Current and future medical therapies. MedscapeCME Oncology; 2009. Fecha de consulta: septiembre de 2010. Disponible en http://cme.medscape.com/viewarticle/709466. [ Links ]
11. Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer. 2008;113(Suppl.):1807-43. [ Links ]
12. Balogh K, Patócs A, Hunyady L, Rácz K. Menin dynamics and functional insight: Take your partners. Mol Cell Endocrinol. 2010;326:80-4. [ Links ]
13. Fainstein P, Fernández T, Mocetti E, Santino J, Balzaretti M, Kozak A, et al. Estudio de la correlación genotipo/fenotipo en la expresión de la menina en el páncreas tumoral y no tumoral de una paciente afectada por neoplasia endocrina múltiple tipo 1 (NEM1). Rev Arg Endocrinol Metab. 2007;4(Supl.):140. [ Links ]
14. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab. 2010;24:355-70. [ Links ]
15. Warner J, Epstein M, Sweet A, Singh D, Burgess J, Stranks S, et al. Genetic testing in familial isolated hyperparathyroidism: Unexpected results and their implications. J Med Genet. 2004;41:155-60. [ Links ]
16. Tsukada T, Nagamura Y, Ohkura N. MEN1 gene and its mutations: Basic and clinical implications. Cancer Sci. 2008 [Epub ahead of print]. [ Links ]
17. Wu X, Hua X. Menin, histone H3 methyltransferases, and regulation of cell roliferation: Current knowledge and perspective. Curr Mol Med. 2008;8:805-15. [ Links ]
18. Dreijerink K, Lips C. Diagnosis and management of multiple endocrine neoplasia type 1 (MEN1). Hered Cancer Clin Pract. 2005;3:1-6. [ Links ]
19. Mejía O. La genética y la medicina predictiva. Implicaciones epistemológicas y consideraciones bioéticas. Acta Med Colomb. 2005;30:126-32. [ Links ]
20. Lips J, Lentjes G, Höppener W. Espectro de tumores y síndromes carcinoides. Acta Bioquím Clín Latinoam. 2004;38:47-59. [ Links ]
21. Yang Y, Hua X. In search of tumor suppressing functions of menin. Mol Cell Endocrinol. 2007;265-266;34-41. [ Links ]
22. Schnepp RW, Chen YX, Wang H, Cash T, Silva A, Diehl JA, et al. Mutation of tumor suppressor gene MEN1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006;66:5707-15. [ Links ]
23. Corbo V, Dalai I, Scardoni M, Barbi S, Beghelli S, Bersani S, et al. MEN1 in pancreatic endocrine tumors: Analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr Relat Cancer. 2010;17:771-83. [ Links ]
24. La P, Yang Y, Karnik SK, Silva AC, Schnepp RW, Kim SK, et al. Menin-mediated caspase 8 expression in suppressing multiple endocrine neoplasia type 1. J Biol Chem. 2007;282:31332-40. [ Links ]
25. Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003;40:657-63. [ Links ]
26. Elaraj DM, Clark OH. Current status and treatment of primary hyperparathyroidism. Perm J Winter. 2008;12:32-37. [ Links ]
27. Kihara M, Miyauchi A, Ito Y, Yoshida H, Miya A, Kobayashi K, et al. MEN1 gene analysis in patients with primary hyperparathyroidism: 10-year experience of a single institution for thyroid and parathyroid care in Japan. Endocr J. 2009;56:649-56. [ Links ]
28. Howell V, Cardinal J, Richardson A, Marsh D. Rapid mutation screening for HRPT2 and MEN1 mutations associated with familial and sporadic primary hyperparathyroidism. J Mol Diagn. 2006;8:559-66. [ Links ]
29. Kandil E, Alabbas HH, Lum YW, Tufaro AP. Familial isolated primary hyperparathyroidism with double adenoma. South Med J. 2010;103:236-8. [ Links ]
30. Mizusawa N, Uchino S, Iwata T, Tsuyuguchi M, Suzuki Y, Mizukoshi T, et al. Genetic analyses in patients with familial isolated hyperparathyroidism and hyperparathyroidism-jaw tumour syndrome. Clin Endocrinol (Oxford). 2006;65:9-16. [ Links ]
31. Simonds W, James-Newton L, Agarwal S, Yang B, Skarulis M, Hendy G, et al. Familial isolated hyperparathyroidism. Clinical and genetic characteristics of 36 kindreds. Medicine. 2002;81:1-26. [ Links ]
32. Teh BT, Farnebo F, Twigg S, Höög A, Kytölä S, Korpi-Hyövälti E, et al. Familial isolated hyperparathyroidism maps to the hyperparathyroidism-jaw tumor locus in 1q21-q32 in a subset of families. J Clin Endocrinol Metab. 1998;83:2114-20. [ Links ]
33. Sharretts JM, Simonds WF. Clinical and molecular genetics of parathyroid neoplasms. Best Pract Res Clin Endocrinol Metab. 2010;24:491-502. [ Links ]
34. Westin G, Björklund P, Akerström G. Molecular genetics of parathyroid disease. World J Surg. 2009;33:2224-33. [ Links ]
35. Georgitsi M, Raitila A, Karhu A, van der Luijt RB, Aalfs CM, Sane T, et al. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab. 2007;92:3321-5. [ Links ]
36. Haase M, Anlauf M, Schott M, Schinner S, Kaminsky E, Scherbaum WA, et al. A new mutation in the menin gene causes the multiple endocrine neoplasia type 1 syndrome with adrenocortical carcinoma. Endocrine. 2010 Nov 11. [Epub ahead of print] [ Links ]
37. Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitors genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. 2009;94:1826-34. [ Links ]
38. Igreja S, Chahal HS, Akker SA, Gueorguiev M, Popovic V, Damjanovic S, et al. Assessment of p27 (cyclin-dependent kinase inhibitor 1B) and aryl hydrocarbon receptor-interacting protein (AIP) genes in multiple endocrine neoplasia (MEN1) syndrome patients without any detectable MEN1 gene mutations. Clin Endocrinol (Oxford). 2009;70:259-64. [ Links ]
39. Pellegata NS, Quintanilla-Martínez L, Siggelkow H, Samson E, Bink K, Höfler H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA. 2006;103:15558-63. [ Links ]
40. Marinoni I, Pellegata NS. p27kip1: A new multiple endocrine neoplasia gene? Neuroendocrinology. 2010 Oct 27. [Epub ahead of print] [ Links ]
41. Molatore S, Marinoni I, Lee M, Pulz E, Ambrosio MR, degli Uberti EC, et al. A novel germline CDKN1B mutation causing multiple endocrine tumors: Clinical, genetic and functional characterization. Hum Mutat. 2010;31:E1825-35. [ Links ]
42. Pigny P, Cardot-Bauters C. Genetics of pheochromocytoma and paraganglioma: new developments. Ann Endocrinol (Paris). 2010;71:76-82. [ Links ]
43. Filocamo M, Regis S, Mazzotti R, Parenti G, Stroppiano M, Gatti R. A simple non-isotopic method to show pitfalls during mutation analysis of the glucocerebrosidase gene. J Med Genet 2001;38:e34. [ Links ]
44. Falchetti A. Genetic screening for multiple endocrine neoplasia syndrome type 1 (MEN-1): When and how. F1000 Med Rep. 2010;2.pii:14. [ Links ]
45. McDermott U, Downing JR, Stratton MR. Genomics and the continuum of cancer care. N Engl J Med. 2011;364:335-40.
[ Links ]
