










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Colombiana de Cirugía
versão impressa ISSN 2011-7582
rev. colomb. cir. vol.30 no.2 Bogotá avr./jun. 2015
1 Médico, posgrado de Cirugía General, Universidad Internacional del Ecuador-Hospital Metropolitano, Quito, Ecuador.
2 Médico cirujano, Servicio de Cirugía General, Hospital Enrique Garcés.
3 Médico nefrólogo, Servicio de Medicina Interna, Departamento de Nefrología, Hospital Metropolitano, Quito, Ecuador.
4 Médico cirujano, Servicio de Cirugía General; jefe, Departamento de Trasplantes, Hospital Metropolitano, Quito, Ecuador.
Correspondencia: Andrés Ayala, MD., Departamento de Cirugía, Hospital Metropolitano, Quito, Ecuador. Correo electrónico: andres_pojkar@hotmail.com
Fecha de recibido: 24 de noviembre de 2014. Fecha de aprobación: 16 de febrero de 2015.
Resumen
En la actualidad, la cirugía hepática y la de la vía biliar son unos de los procedimientos más practicados a nivel mundial. El refinamiento en las técnicas quirúrgicas, los cuidados anestésicos y el trasplante hepático han revolucionado el tratamiento de enfermedades hepáticas que inicialmente eran incurables. La enfermedad poliquística hepática autosómica dominante es rara y puede asociarse a poliquistosis renal o presentarse de forma aislada. Su sintomatología se presenta en los estados avanzados de la enfermedad. A pesar de las múltiples modalidades de tratamiento para esta condición, el manejo quirúrgico con hepatectomía y fenestración es el que ha mostrado mejores resultados en los pacientes con saciedad temprana y hepatomegalia masiva.
Se hace una revisión bibliográfica y se presenta una serie de casos atendidos con esta enfermedad en el Hospital Metropolitano de Quito, Ecuador.
Palabras clave: hígado; poliquístico; riñón poliquístico autosómico dominante; hepatectomía.
Abstract
Nowadays liver and biliary tract surgery are the most commonly performed procedures worldwide; refinement in surgical techniques, anesthetic care and liver transplantation have revolutionized the treatment of liver diseases that were previously incurable. The autosomal dominant polycystic liver disease is a rare condition that may be associated with polycystic kidney disease or can present alone; the symptoms of this condition occur in the advanced stages of the disease. Despite the multiple modalities of treatment available, surgery with hepatectomy and fenestration has shown better results in patients with early satiety and massive hepatomegaly.
A literature review was carried out and a number of cases dealt with this disease at the Metropolitan Hospital in Quito, Ecuador, are presented.
Key words: liver; polycystic; polycystic kidney, autosomal dominant; hepatectomy.
Introducción
En 1856, Bristowe describió por primera vez la asociación de poliquistosis hepática con la enfermedad poliquística renal autosómica dominante en el adulto 1. Esta rara enfermedad tiene una prevalencia de 0,13 a 0,52 % en la población general 2,3.
Se han descrito dos entidades patológicas asociadas con el desarrollo de poliquistosis hepática: la enfermedad poliquística renal autosómica dominante y la enfermedad poliquística hepática autosómica dominante, ambas con un origen genético distinto pero con un desarrollo patológico similar 4; la principal diferencia es la ausencia de quistes renales en la segunda.
La enfermedad poliquística renal autosómica dominante es la enfermedad renal más comúnmente heredada, con un patrón de herencia dominante que se desarrolla por mutaciones en el gen PKD1 (Polycystic Kidney Disease 1) en el 85 % de los casos y el gen PKD2 (Polycystic Kidney Disease 2) en el 15 % restante 5,6.
Esta enfermedad se caracteriza por el desarrollo y crecimiento progresivo bilateral de quistes que surgen del sistema colector renal 7,8, que conduce a un estadio terminal renal; hasta 5 % de estos pacientes requerirán tratamiento de reemplazo renal 9.
La poliquistosis hepática es la manifestación extrarrenal más común de la enfermedad poliquística renal autosómica dominante, con una prevalencia de 58 % entre los 15 y 24 años, que se incrementa hasta 94 % a los 35 a 46 años 10. Aunque es inusual el desarrollo de insuficiencia hepática en la enfermedad poliquística renal autosómica dominante, la afectación quística hepática es responsable de una morbilidad y mortalidad significativas 11. La afectación hepática es más extensa en las mujeres que han tenido varios embarazos o con exposición previa a hormonas sexuales femeninas 12.
Enfermedad poliquística hepática aislada
Generalidades y factores genéticos
En 1952, Comfort, et al., describieron la poliquistosis hepática aislada, en 8 de 24 autopsias 13. En estudios posteriores, identificaron los genes involucrados en el desarrollo de esta enfermedad, el PRKCSH (Protein Kinase C Substrate 80K-H) y el SEC63 (SEC63 homolog- S. cerevisiae) 14-16.
Epidemiología
La enfermedad poliquística hepática aislada se considera rara, con una incidencia menor de 0,01 % 3,17,18. En esta enfermedad los quistes comienzan a aparecer después de la pubertad; las tasas de crecimiento de los quistes y del hígado, son heterogéneas, y los quistes aumentan en número y tamaño progresivamente con la edad 12,19.
Fisiopatología
Los estudios experimentales y en humanos, sugieren varios mecanismos involucrados en la formación de los quistes hepáticos: 1) aumento de la proliferación celular y la apoptosis, 2) secreción abundante de líquido, 3) interacciones anormales entre la célula y la matriz celular, 4) alteraciones de la polaridad celular, y 5) anormalidad en la función estructural ciliar 20.
Los quistes hepáticos se desarrollan en malformaciones de la placa embriológica de los conductos biliares intralobulares, los llamados "complejos de von Meyenburg", 21 los cuales crecen de forma progresiva hasta producir la pérdida de la continuidad en el árbol biliar 22,23.
El hamartoma biliar, descrito por von Meyenburg en 1918, forma parte de las malformaciones de la placa ductal en una fase tardía de su desarrollo, cuando se están formando los conductos biliares periféricos interlobulares y pequeños conductos biliares dilatados, a veces de aspecto quístico, delimitados por un epitelio y rodeados de tejido fibroso.
Cuadro clínico
Hasta en 80 % de los pacientes con esta enfermedad no se presentan síntomas; cuando se presentan, son resultado del efecto de masa de los quistes en estructuras adyacentes o de sus complicaciones. La sintomatología incluye: disnea, saciedad temprana, pesadez, reflujo gastroesofágico, dolor lumbar, edema de miembros inferiores, hepatomegalia, (figura 1), obstrucción biliar y ascitis 23,24.
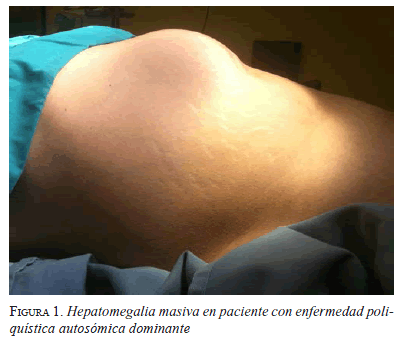
Se han detectado quistes hepáticos aislados en 18 % de las ultrasonografías o tomografías computadorizadas abdominales practicadas por alguna otra enfermedad 24-26. Este hallazgo es cada vez más frecuente, con el uso habitual de estos estudios de imaginología (figura 2) 27.
Exámenes diagnósticos complementarios
En pacientes sintomáticos, las pruebas de función hepática pueden elevarse en forma variable: hasta en 70 %, la gamma-glutamil-transpeptidasa (GGT), hasta en 47 % la fosfatasa alcalina, hasta en 27 % la aspartato aminotransferasa (AST) y hasta en 15 % las bilirrubinas 28-30.
La clasificación más utilizada para la enfermedad poliquística hepática es la tomográfica, teniendo en cuenta el número y tamaño de los quistes y la cantidad de hígado sano residual (tabla 1) 31.

Tratamiento
Hasta el momento actual, ningún tratamiento médico no invasivo ha demostrado ser efectivo en prevenir o disminuir el crecimiento de los quistes en esta enfermedad. Entre las medidas invasivas para los pacientes sintomáticos se mencionan: la aspiración percutánea del quiste seguida de escleroterapia, la fenestración del quiste, la resección hepática con fenestración, la 'embolización' selectiva de la arteria hepática, la colocación de endoprótesis biliares o vasculares y, rara vez, el trasplante de hígado. La elección del tratamiento invasivo específico depende de la cantidad, el tamaño y la localización de los quistes 32. Los objetivos principales del tratamiento quirúrgico se encaminan a reducir el tamaño del hígado sin comprometer su función, además de proporcionar un alivio sintomático a largo plazo 33.
Métodos invasivos de tratamiento
Aspiración y escleroterapia
La aspiración seguida de escleroterapia está indicada en pacientes sintomáticos con grandes quistes hepáticos (mayores de 5 cm) o quistes dominantes. Con el uso de esta técnica, van Keimpema observó una reducción del 17,1 % del volumen hepático medio, en casos de quistes hepáticos aislados grandes y de 19,2 % en aquellos con poliquistosis hepática 27.
Fenestración del quiste
La fenestración del quiste por vía laparoscópica o por laparotomía abierta, implica el destechado y la ablación del área quística 34. Con esta técnica se ha observado un alivio de los síntomas hasta en 80 % de los pacientes, especialmente en aquellos con pocos quistes grandes y superficiales 33, 35-39. Con el abordaje laparoscópico se ha demostrado una corta estancia hospitalaria y baja mortalidad, pero con recurrencia en 50 % de los casos, conversión a cirugía abierta en 10 % y complicaciones como sangrado, infección, pérdida de bilis y ascitis (0-69 %) y mortalidad en 30 % 40,41.
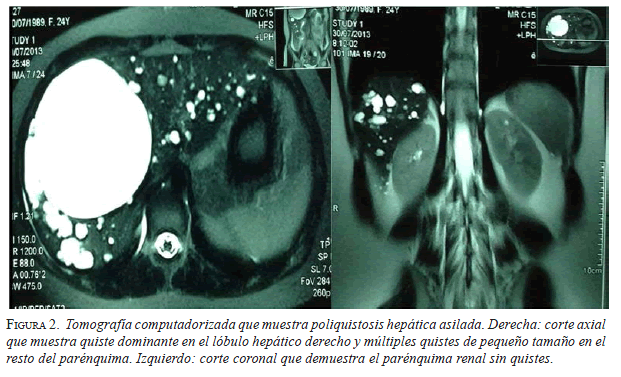
Hepatectomía combinada con fenestración
La resección hepática con fenestración (figura 3) es el tratamiento más eficaz para disminuir el volumen hepático. Esta técnica se usa en pacientes con hepatomegalia masiva por innumerables quistes de pequeño y mediano tamaño, conservando al menos dos segmentos hepáticos contiguos con el drenaje venoso hepático adecuado 32. En la serie de la Clínica Mayo, las tasas de morbilidad y mortalidad fueron de 63 % y 3 %, respectivamente, siendo las complicaciones más comunes la fuga de bilis y la ascitis 38.
Cirugía de trasplante
El trasplante hepático se reserva para los pacientes gravemente sintomáticos en quienes otras modalidades de tratamiento no son adecuadas. Puede tener una mortalidad de hasta 20 % dentro de los primeros seis meses, pero con resultados excelentes a largo plazo en los que sobrevivieron más de este tiempo 33. En un estudio se encontró una tasa de supervivencia al primero, segundo y quinto año de 78,1 %, 71,7 % y 68,7 %, respectivamente 42.
'Embolización'
La 'embolización' selectiva de la arteria hepática ha demostrado una reducción media de 23 % del volumen hepático, con pocos efectos secundarios; se recomienda en pacientes que no toleran el procedimiento quirúrgico 43,44.
Métodos no invasivos de tratamiento
Además de hacerlo en métodos invasivos de tratamiento, varios estudios se han enfocado en identificar las vías implicadas en el desarrollo y el crecimiento de los quistes, con el fin de crear nuevas estrategias de tratamiento. En ratones se ha observado que el octreótido, un análogo de la somatostatina, produce una reducción en la producción del AMPc (3'-5'-monofosfato cíclico de adenosina) en los colangiocitos que recubren el quiste, disminuyendo el volumen hepático en el 19 % y, el volumen de los quistes, en el 40 % 45. Después de este estudio, varios son los ensayos clínicos de asignación aleatoria en que se ha demostraron que el tratamiento con análogos de la somatostatina (octreótido y lanreótido) disminuye el volumen del hígado en un rango de 15 a 38 % 46-50.
Los inhibidores de la proteína mTOR (Mammalian Target of Rapamycin) son inmunosupresores que tienen un efecto antiproliferativo y antiangiogénico, los cuales han demostrado reducir drásticamente el volumen de los quistes, en varios estudios experimentales 51- 55. Sin embargo, los resultados del tratamiento médico siguen siendo inferiores frente al manejo quirúrgico.
A pesar de las múltiples modalidades de tratamiento para esta enfermedad, su selección se hace con base en la anatomía hepática, la reserva hepática, la gravedad de los síntomas y el estado de salud del paciente 38.
Experiencia en el Hospital Metropolitano (Quito, Ecuador)
En nuestra institución, entre 2008 y 2012, se atendieron doce pacientes con enfermedad poliquística hepática, renal o ambas (tabla 2), de los cuales solo 2 (16,6 %) tuvieron enfermedad poliquística hepática aislada. La edad promedio el momento del diagnóstico fue de 51±11 años y el 83,3 % de los pacientes refería antecedentes familiares de afectación poliquística renal o hepática o ambas.
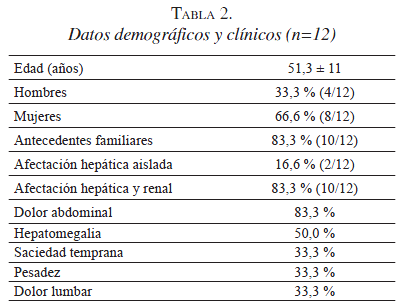
En los pacientes con afectación hepática, el síntoma más frecuente fue el dolor abdominal (83 %), seguido por hepatomegalia (figuras 1 y 2). La hepatectomía por cirugía abierta combinada con la fenestración, fue el procedimiento de elección en pacientes con saciedad temprana y hepatomegalia masiva (figura 3).
Conclusiones
La enfermedad poliquística hepática aislada es una enfermedad poco frecuente, en la cual el tratamiento quirúrgico está indicado para los pacientes sintomáticos; el procedimiento varía de acuerdo con el tamaño y la extensión de la afectación hepática, y puede ir desde la fenestración hasta la resección hepática anatómica.
El conocimiento actual sobre la cirugía hepática y el trasplante hepático, ha permitido tener buenos resultados en el tratamiento de esta enfermedad. La experiencia inicial en nuestra institución con esta enfermedad, ha mostrado buenos resultados con la implementación de hepatectomía por cirugía abierta combinada con la fenestración.
Bibliografía
1. Bristowe F. Cystic disease of the liver associated with similar disease of the kidneys. Trans Pathol Soc Lond. 1856;7:229-34. [ Links ]
2. Kwok M, Lewin K. Massive hepatomegaly in adult polycystic liver disease. Am J Surg Pathol. 1988;12:321-4. [ Links ]
3. Melnick P. Polycystic liver; analysis of seventy cases. Arch Pathol. 1955;59:162-72. [ Links ]
4. Qian Q, Li A, King B, Kamath P, Lager D, Huston J 3rd, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37:164-71. [ Links ]
5. Torres V, Harris P, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287-301. [ Links ]
6. Peters D, Sandkuijl L. Genetic heterogeneity of polycystic kidney disease in Europe. Contrib Nephrol. 1992;97:128-39. [ Links ]
7. Heggo O. A microdissection study of cystic disease of the kidneys in adults. J Pathol Bacteriol. 1966;91:311-5. [ Links ]
8. Verani R, Silva F. Histogenesis of the renal cysts in adult (autosomal dominant) polycystic kidney disease: A histochemical study. Mod Pathol. 1988;1:457-63. [ Links ]
9. Grantham J. Clinical practice: Autosomal dominant polycystic kidney disease. N Engl J Med. 2008;359:1477-85. [ Links ]
10. Bae K, Zhu F, Chapman A, Torres V, Grantham J, Guay-Woodford L, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal dominant polycystic kidney disease: The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1:64-9. [ Links ]
11. Grünfeld J, Albouze G, Jungers P, Landais P, Dana A, Droz D, et al. Liver changes and complications in adult polycystic kidney disease. Adv Nephrol Necker Hosp. 1985;14:1-20. [ Links ]
12. Gabow P, Johnson A, Kaehny W, Manco-Johnson M, Duley I, Everson G. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11:1033-7. [ Links ]
13. Comfort M, Gray H, Dahlin D, Whitesell F Jr. Polycystic disease of the liver: A study of 24 cases. Gastroenterology. 1952;20: 60-78. [ Links ]
14. Drenth JP, Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat.Genet. 2003;33:345-7. [ Links ]
15. Dávila S, Furu L, Gharavi A, Tian X, Onoe T, Qian Q, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575-7. [ Links ]
16. Li A, Dávila S, Furu L, Qian Q, Tian X, Kamath P, et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003;72:691-703. [ Links ]
17. Feldman M. Polycystic disease of the liver. Am J Gastroenterol. 1958;29:83-6. [ Links ]
18. Karhunen P, Tenhu M. Adult polycystic liver and kidney diseases are separate entities. Clin Genet. 1986;30:29-37. [ Links ]
19. Everson GT, Scherzinger A, Berger-Leff N, Reichen J, Lezotte D, Manco-Johnson M, et al. Polycystic liver disease: Quantitation of parenchymal and cyst volumes from computed tomography images and clinical correlates of hepatic cysts. Hepatology. 1988;8:1627-34. [ Links ]
20. Onori P, Franchitto A, Mancinelli R, Carpino G, Alvaro D, Francis H, et al. Polycystic liver diseases. Dig Liver Dis. 2010;42:261-71. [ Links ]
21. Redston M, Wanless I. The hepatic von Meyenburg complex: Prevalence and association with hepatic and renal cysts among 2843 autopsies corrected. Mod Pathol. 1996 9:233-37. [ Links ]
22. Kida T, Nakanuma Y, Terada T. Cystic dilatation of peribiliary glands in livers with adult polycystic disease and livers with solitary nonparasitic cysts: An autopsy study. Hepatology. 1992;16:334-40. [ Links ]
23. Lazaridis K, Strazzabosco M, Larusso N. The cholangiopathies: Disorders of biliary epithelial. Gastroenterology. 2004;127:1565-77. [ Links ]
24. Gaines P, Sampson M. The prevalence and characterization of simple hepatic cysts by ultrasound examination. Br J Radiol. 1989;62:335-7. [ Links ]
25. Caremani M, Vincenti A, Benci A, Sassoli S, Tacconi D. Ecographic epidemiology of non-parasitic hepatic cysts. J Clin Ultrasound. 1993;21:115-8. [ Links ]
26. Carrim Z, Murchison J. The prevalence of simple renal and hepatic cysts detected by spiral computed tomography. Clin Radiol. 2003;58:626-9. [ Links ]
27. van Keimpema L, de Koning D, Strijk S, Drenth J. Aspiration-sclerotherapy results in effective control of liver volume in patients with liver cysts. Dig Dis Sci. 2008;53:2251-7. [ Links ]
28. van Erpecum K, Janssens A, Terpstra J, Tjon A Tham RT. Highly symptomatic adult polycystic disease of the liver. A report of fifteen cases. J Hepatol. 1987;5:109-17. [ Links ]
29. Que F, Nagorney D, Gross J, Torres V. Liver resection and cyst fenestration in the treatment of severe polycystic liver disease. Gastroenterology. 1995;108:487-94. [ Links ]
30. Kabbej M, Sauvanet A, Chauveau D, Farges O, Belghiti J. Laparoscopic fenestration in polycystic liver disease. Br J Surg. 1996;83:1697-701. [ Links ]
31. Gigot J, Jadoul P, Que F, van Beers B, Etienne J, Horsmans Y, et al. Adult polycystic liver disease: Is fenestration the most adequate operation for long-term management? Ann Surg. 1997;225:286-94. [ Links ]
32. Qian Q. Isolated polycystic liver disease. Adv Chronic Kidney Dis. 2010;17:181-9. [ Links ]
33. Russell R, Pinson W. Surgical management of polycystic liver disease. World J Gastroenterol. 2007;13:5052-9. [ Links ]
34. Lin T, Chen C, Wang S. Treatment of non-parasitic cystic disease of the liver: A new approach to therapy with polycystic liver. Ann Surg. 1968;168:921-7. [ Links ]
35. Farges O, Bismuth H. Fenestration in the management of polycystic liver disease. World J Surg. 1995;19:25-30. [ Links ]
36. Konstadoulakis M, Gomatos I, Albanopoulos K, Alexakis N, Leandros E. Laparoscopic fenestration for the treatment of patients with severe adult polycystic liver disease. Am J Surg. 2005;189:71-5. [ Links ]
37. Martin I, McKinley A, Currie E, Holmes P, Garden O. Tailoring the management of nonparasitic liver cysts. Ann Surg. 1998;228:167-72. [ Links ]
38. Schnelldorfer T, Torres V, Zakaria S, Rosen C, Nagorney D. Polycystic liver disease: A critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann Surg. 2009;250:112-8. [ Links ]
39. Manterola C, Pineda V, Vial M. Efectividad del tratamiento laparoscópico de quistes y tumores hepáticos. Revisión global de la evidencia. Revista Chilena de Cirugía. 2007;59:264-71. [ Links ]
40. Robinson T, Stiegmann G, Everson G. Laparoscopic palliation of polycystic liver disease. Surg Endosc. 2005;19:130-2. [ Links ]
41. Hazbón H. Enfermedad poliquística del hígado. Rev Colomb Cir. 2008;23:168-73. [ Links ]
42. US Scientific Registry of Transplant: 2004 OPTN / SRTR Annual Report. US Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients. U.S. Government Sponsored Work, 2004. Fecha de consulta: 23 de septiembre de 2014. Disponible en: www.ustransplant.org [ Links ]
43. Takei R, Ubara Y, Hoshino J, Higa Y, Suwabe T, Sogawa Y, et al. Percutaneous transcatheter hepatic artery embolization for liver cysts in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2007;49:744-52. [ Links ]
44. Ubara Y, Takei R, Hoshino J, Tagami T, Sawa N, Yokota M, et al. Intravascular embolization therapy in a patient with an enlarged polycystic liver. Am J Kidney Dis. 2004;43:733-8. [ Links ]
45. Masyuk T, Masyuk A, Torres V, Harris P, Larusso N. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3',5'-cyclic monophosphate. Gastroenterology. 2007;132:1104-16. [ Links ]
46. van Keimpema L, de Man R, Drenth J. Somatostatin analogues reduce liver volume in polycystic liver disease. Gut. 2008;57:1338-9. [ Links ]
47. van Keimpema L, Drenth J. Effect of octreotide on polycystic liver volume. Liver Int. 2010;30:633-34. [ Links ]
48. van Keimpema L, Nevens F, Vanslembrouck R, van Oijen M, Hoffmann A, Dekker H, et al. Lanreotide reduces the volume of polycystic liver: A randomized, double-blind, placebo-controlled trial. Gastroenterology. 2009;137:1661-8. [ Links ]
49. Hogan M, Masyuk T, Page L, Kubly V, Bergstralh E, Li X, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010;21:1052-61. [ Links ]
50. Caroli A, Antiga L, Cafaro M, Fasolini G, Remuzzi A, Remuzzi G, et al. Reducing polycystic liver volume in ADPKD: Effects of somatostatin analogue octreotide. Clin J Am Soc Nephrol. 2010;5:783-9. [ Links ]
51. Shillingford J, Murcia N, Larson C, Low S, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA. 2006;103:5466-71. [ Links ]
52. Wahl P, Serra A, Le H, Molle K, Hall M, Wuthrich R. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant. 2006;21:598-604. [ Links ]
53. Tao Y, Kim J, Schrier R, Edelstein C. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol. 2005;16:46-51. [ Links ]
54. Wu M, Wahl P, Le H, Wackerle-Men Y, Wuthrich R, Serra A. Everolimus retards cyst growth and preserves kidney function in a rodent model for polycystic kidney disease. Kidney Blood Press Res. 2007;30:253-9. [ Links ]
55. Wu M, Arcaro A, Varga Z, Vogetseder A, Le HM, Wüthrich R, et al: Pulse mTOR inhibitor treatment effectively controls cyst growth but leads to severe parenchymal and glomerular hypertrophy in rat polycystic kidney disease. Am J Physiol Renal Physiol. 2009;297:1597-1605. [ Links ]

{kind=link}
{kind=link}