










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cirugía
Print version ISSN 2011-7582
rev. colomb. cir. vol.32 no.4 Bogotá Oct./Dec. 2017
https://doi.org/10.30944/20117582.38
Doi: https://doi.org/10.30944/20117582.38
1 Fellow de Coloproctología, Universidad del Rosario; cirujano de Cáncer Colorrectal, Universidad de Chile; asesoría genética en Cáncer Heredofamiliar, Universidad de Chile; cirugía general, Universidad Pontificia Bolivariana; Servicio de Cirugía Colorrectal, Hospital Pablo Tobón Uribe, Medellín, Colombia.
1 Cirujano general, Universidad de Antioquia; medicina general, Universidad Industrial de Santander; Servicio de Cirugía General, Hospital San Vicente de Paúl, Medellín, Colombia.
2 Fellow de Coloproctología, Universidad del Rosario; médico epidemiólogo, Universidad del Rosario; cirugía general, Universidad del Rosario; medicina general, Universidad del Rosario, Bogotá, D.C., Colombia.
2 Fellow de Coloproctología, Universidad del Rosario; cirugía general, Universidad Industrial de Santander; medicina general, Universidad Autónoma de Santander, Bucaramanga, Colombia.
Correspondencia: Cristina Isabel Martínez, Medellín, Colombia. cristinamartinezcx@gmail.com
Citar como: Martínez CI, Pérez LF, Baquero D, Barco A. Cáncer colorrectal hereditario no asociado a poliposis o síndrome de Lynch. Rev Colomb Cir. 2017;32:297-303.
Fecha de recibido: 17 de mayo de 2017. Fecha de aprobación: 10 de julio de 2017.
Resumen
El cáncer colorrectal hereditario no asociado a poliposis, también llamado síndrome de Lynch, es reconocido como un síndrome hereditario de patrón autosómico dominante de penetrancia incompleta, en el cual hay mutación en los genes reparadores del ADN.
De 2 a 3 % de todos los tumores colorrectales se originan por este síndrome hereditario que predispone a su desarrollo. El síndrome Lynch, el más frecuente de los síndromes genéticos, incrementa, además del riesgo de desarrollar cáncer de colon, el de cáncer metacrónico y otros tipos de cáncer no colorrectal como los de endometrio, de intestino delgado, de uréter o de la pelvis renal.
Por lo tanto, es indispensable reconocerlo e identificar a los individuos en riesgo de presentarlo para prevenir, diagnosticar y tratar de manera precoz la aparición de estas neoplasias, y poder disminuir las tasas de morbilidad y mortalidad asociadas.
Palabras clave: neoplasias del colon; neoplasias del recto; síndromes neoplásicos hereditarios; neoplasias colorrectales hereditarias sin poliposis; síndrome de Lynch; prevención primaria.
Abstract
Hereditary nonpolyposis colorectal cancer, also known as Lynch syndrome is recognized as an autosomal dominant hereditary syndrome of incomplete penetrance characterized by mutations in DNA repair genes.
It is the most frequent of all the hereditary syndromes, and increases the likelihood of developing colorectal cancer, thus representing 2-3% of all colorectal cancers (CRC). This syndrome predisposes to metachronous (CRC) and other extracolonic cancers, as endometrium, small bowel, ureter and renal pelvis, among others. Therefore, it is necessary to recognize this syndrome and identify individuals with HNPCRC to prevent, diagnose and provide, if possible, early treatment in an effort to decrease its morbidity and mortality.
Key words: Colonic neoplasms; rectal neoplasms; neoplastic syndromes, hereditary; colorectal neoplasms, hereditary nonpolyposis; Lynch syndrome; primary prevention.
Introducción
Según datos publicados en 2012 por la International Agency for Research on Cancer (IARC), a nivel mundial, el colorrectal es el tercer cáncer más común en hombres (10,0 % del total) y el segundo en mujeres (9,2 % del total), y corresponde a la quinta causa de muerte por neoplasias malignas, con una mortalidad baja, de 9,8 %. El 55 % de los casos de cáncer colorrectal ocurrieron en países con un alto índice de desarrollo, y se estima que su incidencia en la población mundial podría aumentar el 75 % en el año 2030. Según el índice de desarrollo humano, los países más ricos tendrían el 40 % de la incidencia global y, los países más pobres, una cercana al 15 % 1. La mortalidad por esta enfermedad ha tenido una disminución sostenida cercana al 50 % en los últimos años, con respecto a las tasas pico de mortalidad previamente reportadas 2.
Se estima que en 20 a 30 % de los pacientes con cáncer colorrectal existe historia familiar de cáncer y, con frecuencia, los familiares de primer grado de pacientes con adenomas colorrectales o cáncer colorrectal tienen mayor riesgo de desarrollar este tipo de tumor, lo cual sugiere un componente hereditario; sin embargo, solo en 5 % de los casos se logra identificar un patrón hereditario bien establecido o un síndrome genético, como síndrome de Lynch, poliposis adenomatosa familiar, poliposis asociada a MUTYH, poliposis juvenil, poliposis hereditaria mixta o síndrome de Peutz-Jeghers 3,4.
Para identificar los pacientes con riesgo de padecer cáncer hereditario, es fundamental una adecuada anamnesis con descripción e identificación detallada de su grupo familiar, y la caracterización estricta de los integrantes o miembros afectados por cáncer, la edad de presentación y los diferentes órganos afectados por cáncer en su familia 5.
Aldred Warthin, patólogo de la Universidad de Michigan, sentó las bases para la comprensión del carácter hereditario de algunos tipos de cáncer y ha sido nombrado "el padre de la genética del cáncer" 6. En 1913, se publicó su investigación basada en la historia familiar de una de sus empleadas, en cuya familia había varios miembros afectados por cáncer, y a la cual denominó "familia G", considerada una de las genealogías de cáncer más largas y mejor detalladas en el mundo. Este trabajo se considera uno de los primeros en identificar el efecto de la herencia y la expresión fenotípica del cáncer en los seres humanos 5.
A pesar de su extensa investigación y grandes esfuerzos, se necesitaron muchas décadas para que su teoría sobre el carácter hereditario del cáncer fuera aceptada, sustentada en posteriores investigaciones de Henry T. Lynch quien, en 1966, reportó dos familias con cáncer colorrectal hereditario; posteriormente, se reportaron cientos de familias con el mismo patrón hereditario de cáncer a nivel mundial. A principios de la década de 1990, se logró identificar el defecto genético o mutación en los genes reparadores del ADN que producen este síndrome 7.
El avance continuo de la ciencia y la tecnología, el mejor entendimiento molecular y genético del cáncer, la identificación de los genes implicados en los diferentes síndromes, las pruebas genéticas para el diagnóstico, la asociación entre los diferentes tipos de cáncer que han demostrado agregación familiar y proponen una predisposición hereditaria al desarrollo de cáncer, incluyendo los tumores de mama, endometrio, ovario, urotelio, colorrectal y próstata, han mejorado nuestra comprensión de los síndromes hereditarios, en especial, la del cáncer colorrectal hereditario, el riesgo de padecerlo y la patogénesis de la enfermedad, y se han propuesto estrategias para su prevención y diagnóstico precoz 6,7.
En los últimos años, con el descubrimiento e identificación de los genes de predisposición para el desarrollo de cáncer de mama y ovario, BRCA1 en 1994 y BRCA2 un año después 6,8, y la secuenciación del genoma humano en el 2003, se ha enfocado la atención en tratar de identificar a las personas en riesgo, pertenecientes a grupos familiares con historia de cáncer hereditario, para brindarles los beneficios de una adecuada atención.
A pesar de esto, hay pocos registros a nivel mundial que nos acerquen a la realidad sobre la incidencia de los diferentes síndromes familiares y hereditarios causantes de cáncer. Registros aislados, como el registro nacional de familias con síndrome de Lynch en Holanda, promueven la identificación de las familias afectadas y alientan la participación de estos individuos en programas de vigilancia, con lo cual se ha observado una disminución significativa de la mortalidad por cáncer en estas familias 9,10.
Síndrome de Lynch
En 1966, Henry T. Lynch reportó dos familias en las cuales se presentaron varios casos de cáncer colorrectal asociado a tumores de endometrio y de estómago, y los diagnosticó como "síndrome de cáncer familiar". Posteriormente, se cambió su nombre a cáncer colorrectal hereditario no asociado a poliposis, para diferenciarlo de otra entidad previamente conocida como poliposis adenomatosa familiar, la cual es un síndrome hereditario por mutación del gen APC.
En la actualidad, se usa el nombre de síndrome de Lynch solo cuando se trata de la manifestación clínica de la mutación genética confirmada de alguno de los genes reparadores del ADN o pérdida de expresión del gen MSH2 por delación en el gen EpCAM 11. Al antes llamado 'cáncer colorrectal no polipoide hereditario', hoy en día se le denomina 'cáncer colorrectal no asociado a poliposis', cuando se cumplen los criterios de Ámsterdam II pero no se ha confirmado la mutación genética.
El síndrome de Lynch es el fenotipo más conocido de cáncer colorrectal hereditario, la mayoría de los tumores colorrectales se desarrollan en pólipos con histología adenomatosa y no como se pensaba antiguamente, que estos tumores se desarrollaban a partir de lesiones no polipoides 11.
Biología molecular
El síndrome de Lynch es un trastorno autosómico dominante de penetrancia incompleta pero cercana al 80 %. Este síndrome se produce por mutación germinal en uno de los alelos de los genes reparadores del ADN (Mismatch Repair Genes, MMR) como primer evento y una segunda mutación que se produce después del nacimiento en el alelo sano de estos genes (teoría de Knudson o del segundo golpe). En individuos con mutación de alguno de estos genes, el riesgo de padecer cáncer colorrectal o cánceres asociados al síndrome, es de 80 % a lo largo de la vida 12.
En este síndrome, la aparición de los tumores en diferentes órganos se debe a la mutación en la línea germinal de uno de los genes reparadores del ADN (MLH1, MSH2, MSH6 o PMS2, y silenciamiento de MSH2 por mutación en el gen EpCAM), que produce inestabilidad microsatelital, que es otra característica fenotípica de los tumores de este síndrome hereditario.
Los genes reparadores tienen como función mantener y garantizar una copia exacta de la cadena replicada durante la replicación del ADN, mediante la corrección de pequeñas inserciones o deleciones generadas por incorporaciones erróneas o deslizamientos de la polimerasa durante la replicación del ADN, especialmente, en las secuencias repetidas, es decir, los microsatélites; dichos errores o deslizamientos se expresan o traducen, a su vez, como inestabilidad microsatelital. La inestabilidad microsatelital es típica de estos tumores y se encuentra en 90 % de los pacientes con tumores malignos que sufren el síndrome de Lynch y en 12 % de aquellos con tumores esporádicos; en estos casos, los tumores son de mejor pronóstico oncológico 13.
Por otra parte, la alteración en el gen EpCAM (Epithelial Celular Adhesion Molecule) que se encuentra en el extremo de MSH2 produce un silenciamiento de la función reparadora del gen MSH2, que inactiva su función y genera un fenotipo similar al del síndrome de Lynch 13,14.
Las mutaciones de los genes reparadores del ADN (MMR) ocurren con mayor frecuencia en MLH1 y MSH2, los cuales son responsables de cerca del 71 % de las mutaciones en el síndrome de Lynch, mientras que solo 15 % lo son de la mutación en MSH6 y una minoría de la mutación de PMS2 (hasta 14 %) y de la de EpCAM 15.
Asimismo, se pueden producir cambios epigenéticos, como la hipermetilación de promotores, que causan inactivación o silenciamiento de genes y causan una expresión fenotípica similar a la del síndrome de Lynch sin haber mutación de los genes MMR, en cuyo caso se denomina cáncer colorrectal hereditario no asociado a poliposis.
El defecto de los genes reparadores se puede establecer por inmunohistoquímica en el tejido tumoral, usando anticuerpos contra las proteínas reparadoras del ADN: MLH1, MSH2, MSH6 y PMS2. Al demostrarse pérdida de expresión de alguna de las proteínas reparadoras del ADN, se puede dirigir y asistir a la identificación del gen mutado por medio de un estudio genético para confirmar el posible gen afectado, con menor costo por ser un estudio dirigido. No obstante, se debe tener en cuenta que en el 75 % de los tumores colorrectales con pérdida de expresión en la inmunohistoquímica de MLH1, se logra identificar una mutación; en el resto de estos tumores, se encuentra hipermetilación somática del promotor de MLH1 lo que se expresa como inactivación del gen 15.
Inestabilidad microsatelital
Los microsatélites STR (Simple Sequence Repeat, SSR o Short Tamden Repeat) son secuencias de ADN en las que un fragmento se repite de manera consecutiva, y se encuentran en regiones no codificantes del ADN.
La inestabilidad microsatelital se caracteriza por una expansión o contracción anormal de estos microsatélites en el tejido tumoral, pero no en el ADN de la mucosa colorrectal sana adyacente. Se produce por un defecto de la capacidad de reparar los errores cuando se hace una copia del ADN de células de pacientes con mutación en los genes reparadores (mutación germinal) o hipermetilación del promotor del gen MLH1 (cambios somáticos en el gen) 11. La inestabilidad microsatelital se encuentra en 90 % de los tumores en pacientes con síndrome de Lynch y en 12 a 20 % de aquellos con cáncer colorrectal esporádico asociado a hipermetilación somática en el promotor del gen MLH1 11,12.
Presentación clínica
El cáncer colorrectal es la expresión fenotípica mas conocida del síndrome de Lynch. Se estima que el riesgo de desarrollar cáncer colorrectal a lo largo de la vida en los portadores de esta mutación es de 50 a 80 %; lo siguen el riesgo de cáncer de endometrio (40 a 60 %) y el de cáncer en otros órganos, que va a depender del tipo de gen reparador del ADN que esté mutado 16.
Frecuentemente, el cáncer colorrectal en casos de síndrome de Lynch se diagnostica antes de los 50 años de edad, más específicamente entre los 44 y los 61, más precozmente que el cáncer colorrectal esporádico que, en promedio, se diagnostica a los 69 años.
La carcinogénesis acelerada se debe a que la progresión de adenoma a carcinoma es más rápida en el síndrome de Lynch; en promedio, el adenoma progresa a cáncer en 35 meses. En cambio, en el cáncer esporádico, la duración de esta progresión se estima en 10 a 15 años (tabla 1) 17,18.
Tabla 1.
Edad promedio del diagnóstico de cáncer colorrectal
según la mutación en los genes reparadores de ADN.
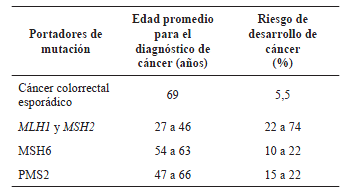
Otra característica fenotípica del síndrome de Lynch es la predilección de los tumores por el colon proximal al ángulo esplénico, reportada en 60 a 80 % de los casos 19. El desarrollo de cáncer colorrectal, sincrónico o metacrónico, es otra característica importante del síndrome de Lynch 18. El riesgo acumulado de tumores metacrónicos por cada 10 años de vida en estos individuos se calcula en 16 % a los 10 años y en 41 % a los 20 años, el cual es directamente proporcional a la aparición de nuevos cánceres en el mismo órgano y en otros órganos afectados según el gen mutado (tabla 2).
Tabla 2.
Riesgo de tumores en órganos diferentes al colon,
según el gen reparador de ADN que ha mutado.
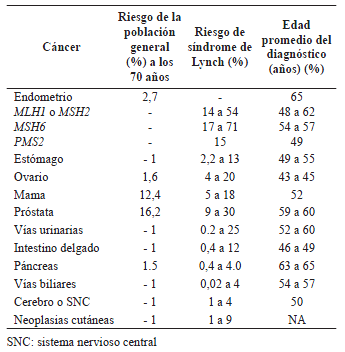
Existen otras características histológicas propias del cáncer colorrectal en el síndrome de Lynch, como la pobre diferenciación de las células tumorales, las células en anillo de sello, el infiltrado linfocitario, los tumores mucoproductores con abundantes acúmulos de mucina y la importante reacción desmoplásica, que permiten su diferenciación de tumores en el mismo estadio correspondientes al cáncer esporádico 19.
Otra de las características del síndrome de Lynch es el mayor riesgo de tumores malignos de origen diferente al colon debido al gen afectado, como el cáncer endometrial que se presenta en 18 a 60 % de las mujeres con mutación del MSH6, cuya edad promedio de diagnóstico es a los 50 años. Otra neoplasia asociada a este síndrome es el carcinoma gástrico, cuyo riesgo se calcula en 6 a 13 %, valor que puede variar cuando es endémico en la población, como ocurre en Corea del Norte, donde puede llegar al 30 % y supera al del cáncer de endometrio 19.
Existen otros subtipos del síndrome de carcinoma colorrectal hereditario no polipoide, como el síndrome de Muir-Torre, en el cual se asocia con carcinoma sebáceo y queratoacantoma, y el síndrome de Turcot, en el cual una neoplasia maligna del sistema nervioso central, como el glioblastoma, se presenta sincrónicamente con adenomas de colon; en ambos síndromes hay mutación de los genes reparadores del ADN. Menos clara es su asociación con el cáncer de mama y el pancreático, aunque algunos investigadores la han informado 19,20.
En 1990, el International Collaborative Group on Hereditary Non-Polyposis Colon Cancer se reunió para unificar conceptos y establecieron los "criterios diagnósticos de Ámsterdam I", para facilitar la identificación de los pacientes con riesgo de ser portadores de las mutaciones causantes del síndrome de Lynch. En 1999, a la luz de nuevos conocimientos sobre los mecanismos genéticos implicados y de la importancia de los tumores asociados no colorrectales, estos criterios se ampliaron y dieron lugar a los hoy llamados criterios de Ámsterdam II, con una sensibilidad de 22 % y una especificidad de 98 % para identificar los pacientes con síndrome de Lynch.
Se usa la nemotecnia "3-2-1" para estos criterios, la cual es: 3 o más familiares con cánceres asociados al síndrome de Lynch y uno de ellos con primer grado de consaguinidad con el afectado; 2 o más generaciones sucesivas afectadas; y 1 o más parientes con cáncer diagnosticado antes de los 50 años; además, que se haya descartado poliposis adenomatosa familiar en casos de cáncer colorrectal 19,21,22 (tabla 3).
Tabla 3.
Criterios de Ámsterdam II para el diagnóstico
de síndrome de cáncer colorrectal hereditario no
asociado a poliposis.

Estudio genético
Se deben ofrecer pruebas genéticas, previa asesoría, a los pacientes: que cumplan con los criterios de Ámsterdam II o con los criterios revisados de Bethesda (Revised Bethesda Guidelines, National Cancer Institute). Si el paciente no satisface los criterios mencionados no se le debe recomendar ofrecer la realización de las pruebas genéticas; si hay inestabilidad microsatelital en pacientes menores de 50 años con tumores de colon, se les pueden ofrecer estas pruebas 21; jóvenes con cáncer asociado al síndrome de Lynch y pérdida de expresión de las proteínas reparadoras del ADN según la inmunohistoquímica; menores de 70 años con pérdida de expresión de proteínas reparadoras del ADN según la inmunohistoquímica; y mayores de 70 años con historia familiar sugestiva de síndrome de Lynch e importante inestabilidad microsatelital 23.
Cuando los estudios genéticos demuestren mutación en los genes asociados al síndrome de Lynch, el paciente debe recibir asesoría genética para evaluar el riesgo individual de otros tumores, según el gen afectado. De esta manera, puede confirmarse el diagnóstico de síndrome de Lynch y, posteriormente, identificarse familiares en riesgo de padecer el síndrome 11.
Vigilancia y seguimiento según la mutación genética
Los pacientes con una mutación confirmada causante del síndrome de Lynch presentan un riesgo elevado de cáncer colorrectal y no colorrectal en edades tempranas. Por lo tanto, se recomienda una evaluación clínica anual, educar al paciente a partir de los 20 a 25 años y a su familia, y practicarse colonoscopia de tamización cada uno o dos años a partir de los 20 a 25 años, o dos a cinco años antes del diagnóstico del individuo más joven con cáncer colorrectal en la familia.
Teniendo en cuenta que la progresión de adenoma a carcinoma es acelerada, se recomienda un intervalo más corto entre las colonoscopias, pues se ha demostrado que la tamización en casos de cáncer colorrectal disminuye la mortalidad por cáncer que es cercana al 72 %, reduce en 62 % la incidencia de cáncer colorrectal y aumenta la expectativa de vida 21.
En todas las mujeres, a partir de los 30 a 35 años, para la tamización y la prevención del cáncer endometrial se debe hacer un examen pélvico anual con biopsia de endometrio y, para las del cáncer de ovario, una ecografía transvaginal anual. La histerectomía y la ooforectomía profiláctica bilateral en mujeres con número satisfecho de partos o en edades cercanas a los 40 años, ha demostrado ser una estrategia eficaz para la prevención del cáncer de ovario o endometrio, la cual disminuye la mortalidad que causan 24.
Para el cribado del cáncer gástrico, está indicada la endoscopia digestiva superior con biopsias después de los 30 años y la erradicación de Helicobacter pylori, con endoscopia de control cada dos a tres años.
Para el diagnóstico temprano de cáncer urotelial debe considerarse el uroanálisis a partir de los 30 a 35 años 24. En la tabla 2 se muestra el riesgo calculado de padecer los diferentes tipos de cáncer, según el gen mutado 11.
Tratamiento
El tratamiento quirúrgico y médico del síndrome de Lynch debe estar dirigido por un grupo multidisciplinario. La colectomía total con anastomosis ileorrectal es el tratamiento de elección para pacientes con síndrome de Lynch y cáncer de colon no susceptible de manejo endoscópico, el cual disminuye en 30 % el riesgo de tumor metacrónico colorrectal a los 10 años.
El riesgo acumulado de presentar cáncer colorrectal después de una colectomía parcial a los 10 años es de 19 %, a los 20 años es de 41 % y a los 30 años es de 62 %. La colectomía parcial o segmentaria es una estrategia quirúrgica que se debe tener en cuenta en pacientes ancianos, en aquellos con disfunción o daño esfinteriano previo y en quienes que hayan recibido radioterapia pélvica.
No se han demostrado diferencias en la calidad de vida obtenida después de colectomía parcial en comparación con la colectomía subtotal, aunque esta última intervención se asoció con peores resultados funcionales en cuanto a la frecuencia de la defecación y el impacto social en estudios iniciales 25. La proctocolectomía total con bolsa ileal y anastomosis ileoanal se reserva para pacientes con el síndrome de Lynch y cáncer de recto, previo manejo según el estadio del cáncer rectal en el momento del diagnóstico 11.
Aunque la evidencia creciente mas no concluyente es a favor del uso de la aspirina en pacientes portadores de la mutación, se debe tener en cuenta y plantearse según una adecuada explicación de los riesgos y beneficios ofrecidos por esta quimioprofilaxis, sobre todo en pacientes con lesiones preneoplásicas previas 26.
Referencias
1. Bray F, Jemal A, Grey N, Ferlay J, Forman D. Global cancer transitions according to the Human Development Index (2008-2030): A population-based study. Lancet Oncol. 2012;13:790-801. [ Links ]
2. Siegel RL, Miller KD, Jemal A. Cancer statistics. Cancer J Clin. 2017;677-30. [ Links ]
3. Quintero E, Carrillo M, Leoz M-L, Cubiella J, Gargallo C, Lanas A, et al. Risk of advanced neoplasia in first-degree relatives with colorectal cancer: A large multicenter cross-sectional study. PLoS Med. 2016;13:e1002008. doi: 10.1371/journal.pmed.1002008. [ Links ]
4. Jansen M, Menko FH, Brosens LAA, Giardiello FM, Offerhaus GJ. Establishing a clinical and molecular diagnosis for hereditary colorectal cancer syndromes: Present tense, future perfect? Gastrointest Endosc. 2014;80:1145-55. [ Links ]
5. McKusick VA. Mendelian inheritance in man and its online version, OMIM. Am J Hum Genet. 2007;80:588-604. [ Links ]
6. Lynch HT. Classics in oncology. Aldred Scott Warthin, M.D., Ph.D. (1866-1931). CA Cancer J Clin. 1985;35:345-7. [ Links ]
7. Vasen HFA, Blanco I, Aktan-Collan K, Gopie JP, Alonso A, Aretz S, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut. 2013;62:812-23. [ Links ]
8. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66-71. [ Links ]
9. Vasen HF, den Hartog Jager FC, Menko FH, Nagengast FM. Screening for hereditary non-polyposis colorectal cancer: A study of 22 kindreds in the Netherlands. Am J Med. 1989;86: 278-81. [ Links ]
10. Järvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomäki P, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829-34. [ Links ]
11. Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US multi-society task force on colorectal cancer. Am J Gastroenterol. 2014;109:1159-79. [ Links ]
12. Boyle TA, Bridge JA, Sabatini LM, Nowak JA, Vasalos P, Jennings LJ, et al. Summary of microsatellite instability test results from laboratories participating in proficiency surveys: Proficiency survey results from 2005 to 2012. Arch Pathol Lab Med. 2014;138:363-70. [ Links ]
13. Ghanipour L, Jirström K, Sundström M, Glimelius B, Birgisson H. Associations of defect mismatch repair genes with prognosis and heredity in sporadic colorectal cancer. Eur J Surg Oncol. 2017;43:311-21. [ Links ]
14. Bhattacharya P, McHugh T. Lynch syndrome. Fecha de consulta: 5 de julio de 2017. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/28613748 [ Links ]
15. Romero-Laorden N, Castro E. Inherited mutations in DNA repair genes and cancer risk. Curr Probl Cancer. 2017;41:251-64. doi.org/10.1016/j.currproblcancer.2017.02.009 [ Links ]
16. Samadder NJ, Jasperson K, Burt RW. Hereditary and common familial colorectal cancer: Evidence for colorectal screening. Dig Dis Sci. 2015;60:734-47. [ Links ]
17. Edelstein DL, Axilbund J, Baxter M, Hylind LM, Romans K, Griffin CA, et al. Rapid development of colorectal neoplasia in patients with Lynch syndrome. Clin Gastroenterol Hepatol. 2011;9:340-3. [ Links ]
18. Colling R, Church DN, Carmichael J, Murphy L, East J, Risby P, et al. Screening for Lynch syndrome and referral to clinical genetics by selective mismatch repair protein immunohistochemistry testing: An audit and cost analysis. J Clin Pathol. 2015;68:1036-9. [ Links ]
19. Wells K, Wise PE. Hereditary colorectal cancer syndromes. Surg Clin North Am. 2017;97:605-25. [ Links ]
20. Flux K. Sebaceous neoplasms. Surg Pathol Clin. 2017;10:367-82. [ Links ]
21. Bui QM, Lin D, Ho W. Approach to Lynch syndrome for the gastroenterologist. Dig Dis Sci. 2017;62:299-304. [ Links ]
22. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453-6. [ Links ]
23. Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11:35-41. [ Links ]
24. Brosens LAA, Offerhaus GJA, Giardiello FM. Hereditary colorectal cancer: Genetics and screening. Surg Clin North Am. 2015;95:1067-80. [ Links ]
25. Kalady MF, Lipman J, McGannon E, Church JM. Risk of colonic neoplasia after proctectomy for rectal cancer in hereditary nonpolyposis colorectal cancer. Ann Surg. 2012;255:1121-5. [ Links ]
26. Burn J, Bishop DT, Mecklin J-P, Macrae F, Möslein G, Olschwang S, et al. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N Engl J Med. 2008;359: 2567-78. [ Links ]

