Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Los tumores neuroendocrinos son un grupo heterogéneo de neoplasias que se originan de las células enterocromafines derivadas de la cresta neural y el endodermo, las cuales se distribuyen en todo el organismo como glándulas o en forma difusa 1. La incidencia en los Estados Unidos para el año 2012 fue de 6,98 casos por 100.000 personas, el 52,7% en mujeres, con un incremento más del 600 % en las últimas cuatro décadas 2. Suelen diagnosticarse entre la sexta y la séptima décadas de la vida. En el 58 % de los casos, el tubo digestivo es la localización primaria de los tumores neuroendocrinos (17 % en el intestino delgado, 17 % en el recto y 6 % en el páncreas), en el 27 % son los pulmones y en el 15 % se desconoce 1.
Entre el 2001 y el 2010 en el Hospital Universitario Fundación Santa Fe de Bogotá, se presentaron 1.221 casos de tumores neuroendocrinos, la mayoría (56,9 %) en mujeres, y el promedio de edad fue de 51,16 años (mediana de 52; rango de 1 a 92). El tumor más frecuente fue el hipofisiario (47 %), seguido del gastrointestinal (15 %), el pulmonar (11 %), el primario desconocido (7 %), el de paratiroides (5 %), el feocromocitoma-paraganglioma (4 %) y el de páncreas (3 %). Con excepción del tumor hipofisiario, todos los tumores neuroendocrinos tienen un potencial maligno considerable y el órgano más comprometido por metástasis es el hígado. En la mayoría de los pacientes, el diagnóstico se hizo por las manifestaciones clínicas. El 61 % de los tumores tenían un el índice de proliferación celular Ki-67 ≤ 3 % y el 90 % fueron bien diferenciados. De los tumores neuroendocrinos carcinoides de pulmón, el 11 % de los típicos y 67 % de los atípicos hicieron metástasis. De los gastroenteropancreáticos, 18 % de los bien diferenciados y el 50 % de los moderadamente diferenciados dieron metástasis. De los tumores neuroendocrinos con primario desconocido, el 31 % se clasificaron como bien diferenciados, y el 9 %, como moderadamente diferenciados 3.
La mayoría de los estos tumores se caracterizan por ser de curso clínico silencioso y de crecimiento lento, compartir la apariencia histológica, y presentar marcadores endocrinos como la cromogranina A, la sinaptofisina y otros 4, los cuales se usan en el diagnóstico en asociación con la clínica y las imágenes diagnósticas 5. Los síntomas están asociados a la secreción de hormonas y a la compresión local por el crecimiento tumoral, las metástasis o ambos.
En general, todos los tumores neuroendocrinos pueden aparecer esporádicamente (mutaciones somáticas encontradas en las células tumorales) o en el contexto de un síndrome genético hereditario (mutaciones germinales encontradas en cualquier célula tumoral y en la célula normal).
Un síndrome genético hereditario se presenta en 5 a 10 % de los casos de tumores neuroendocrinos. Entre las condiciones genéticas implicadas, están: los síndromes de neoplasia endocrina múltiple de tipo 1 (gen MEN-1 o menina), de tipo 2 (gen RET) y de tipo 4 (gen CDKN1B) 6; la enfermedad de von Hippel-Lindau (gen VHL); la neurofibromatosis 1 (gen NF1); la esclerosis tuberosa (genes TSC1 y TSC2); la tríada de Carney (gen no identificado); los síndromes de Carney-Stratakis (genes SDHB, SDHC y SDHD) 4,7, de Reed (gen FH), de policitemia-paraganglioma-somatostatinoma (gen HIF2A) de paraganglioma-feocromocitoma (genes SDHx, SDHAF2, MAX, TMEM127, HIF2A, PHD1, PHD2, HRAS, KIF1B, DH2, KRAS, IDH, FH y BAP-1), y los adenomas pituitarios aislados familiares (gen AIP). Se han reportado otras mutaciones germinales de los genes de inhibidores de ciclina dependientes de cinasas asociadas como: CDKN2B (p15), CDKN2C (p18) y CDKN1A (p21) 8,9.
Los tumores neuroendocrinos de páncreas, previamente conocidos como tumores de los islotes pancreáticos, se desarrollan en las células endodérmicas embrionarias que dan origen a los islotes de Langerhans. Estas células son especializadas y producen, almacenan y secretan péptidos y aminas biogénicas 4.
Por lo tanto, estos tumores pueden secretar una variedad de hormonas peptídicas, incluyendo insulina, gastrina, glucagón, somatostatina, polipéptido pancreático y péptido intestinal vasoactivo, entre otros. Esto resulta en una amplia variedad de síndromes clínicos asociados. Sin embargo, entre 50 y 75 % de los tumores neuroendocrinos de páncreas pueden no ser funcionales y suelen tener una presentación tardía con síntomas inespecíficos, como pérdida de peso, dolor abdominal y anorexia, debido a la secreción de diversos péptidos que no dan un síndrome clínico. Usualmente, en 40 a 50 % de los casos, producen tardíamente síntomas relacionados con compresión local, metástasis o ambas 10; en el porcentaje restante, son asintomáticos y se diagnostican como hallazgos incidentales.
Los tumores neuroendocrinos de páncreas son la segunda neoplasia epitelial más frecuente del páncreas, con una mortalidad del 60% 11; tienen una incidencia menor de 1 caso por 100.000 personas por año y prevalencia del 10 % 4. Los datos del Surveillance, Epidemiology and End Results (SEER) muestra un incremento en la incidencia de 0,17 en 1970 a 0,8 en 2012, con una prevalencia a 20 años de 0,003 % y con la menor supervivencia a cinco años comparado con otros tumores neuroendocrinos gastrointestinales 4.
La mediana de la supervivencia para la enfermedad local es de 230 meses, para la local o regional es de 90 meses y para la metastásica es de 20 meses; para el tumor neuroendocrino de páncreas bien diferenciado es de 140 meses, para el moderadamente diferenciado es de 70 meses y para el mal diferenciado es de 10 meses. Asimismo, los estudios post mortem reportan una incidencia del 0,8 a 10 % 12. Aunque el pico de incidencia ocurre entre los 40 y los 69 años, un número significativo de pacientes se diagnostican antes de los 35 años. La relación hombre a mujer es de 1,4 a 1. El 70 % de los tumores neuroendocrinos funcionales de páncreas corresponden a insulinomas, y el 90 % de estos son benignos; el 15 % son glucagonomas, y el 10 % son gastrinomas o somastotatinomas, los cuales tienen un riesgo de metástasis de 80 a 90 % 1.
Clasificación patológica de los tumores neuroendocrinos
Los tumores neuroendocrinos de páncreas se clasifican, en primer lugar, como funcionales o no funcionales, según los síntomas relacionados con la secreción hormonal tumoral. Generalmente, los que están bien diferenciados, muestran producción hormonal y se asocian con síndromes hormonales reconocidos (insulinoma, gastrinoma, etc.); aquellos pobremente diferenciados raramente se asocian con secreción hormonal.
En las últimas dos décadas, su clasificación histopatológica ha tenido varios cambios relacionados con la comprensión de la enfermedad. En el 2000, se incluía el estadio tumoral (TNM) del American Joint Committee on Cáncer (AJCC) y la clasificación de la Organización Mundial de la Salud (OMS), la cual comprendía tres categorías: carcinoma bien diferenciado con un comportamiento benigno o incierto, carcinoma bien diferenciado con características malignas y carcinoma pobremente diferenciado.
Esta clasificación se actualizó en el 2010, teniendo en cuenta que las características patológicas usadas no se correlacionaban adecuadamente con el pronóstico y el comportamiento biológico de los tumores y, por ende, era imperfecto para determinar el mejor tratamiento 13. Asimismo, se observó que las características histológicas del tumor influían aún más en estos factores. Por este motivo, se decidió dar mayor importancia al grado histológico del tumor y se incluyó en la clasificación el conteo mitótico y el índice de proliferación celular determinado con el marcador Ki-67. Es indispensable corroborar el fenotipo tumoral con estudios de inmunohistoquímica de cromogranina, sinaptofisina o ambas.
Se establecieron tres categorías histológicas según la característica evaluada, el recuento mitótico o el índice de Ki-67, con mayor valor. Por ejemplo, si un tumor neuroendocrino de páncreas es bien diferenciado, con un conteo mitótico menor de 2 y un índice Ki-67 de 2 %, se considera de grado 1; pero si el mismo tumor presenta un índice Ki-67 de 25 %, se considera un carcinoma neuroendocrino de grado 3, lo cual cambia el pronóstico 14.
No obstante, esta clasificación está siendo modificada, en especial, por el conocimiento de los mecanismos moleculares subyacentes a la tumorogénesis neuroendocrina pancreática. En la mayoría de tumores neuroendocrinos bien diferenciados, se identifican alteraciones en la remodelación de la cromatina por la vía PI3K-AKT-mTOR y, en los carcinomas neuroendocrinos pobremente diferenciados, las mutaciones en TP53 y RB pueden contribuir a su desarrollo y ayudan a diferenciarlos 15. Probablemente, estos mecanismos expliquen la discordancia, entre el índice Ki-67, el conteo mitótico y el pronóstico en años de supervivencia, observada en múltiples estudios 14.
Por lo anterior, los últimos estudios plantean la necesidad de establecer dos tipos de grado 3: uno bien diferenciado con Ki-67 alto y otro pobremente diferenciado, basado en la morfología y en los cambios moleculares. Estos tumores neuroendocrinos de grado 3 tienen morfología bien diferenciada con un patrón de crecimiento organoide, no expansivo, ausencia de necrosis geográfica y sin estroma desmoplásico 14,16,17.
En 2017, la OMS actualizó la clasificación histológica de los tumores neuroendocrinos de páncreas de la siguiente manera 18:
Tumores neuroendocrinos bien diferenciados
Tumor neuroendocrino pobremente diferenciado
Neoplasia mixta neuroendocrina y no neuroendocrina
Grado 1:. tumor bien diferenciado, índice Ki-67 menor de 3 % e índice mitótico menor de 2 por 10 campos de mayor aumento
Grado 2:. tumor bien diferenciado, índice Ki-67 de 3 a 20 % e índice mitótico de 2 a 20 por 10 campos de mayor aumento
Grado 3:. tumor bien diferenciado, índice Ki-67 mayor de 20 % e índice mitótico mayor de 20 por 10 campos de mayor aumento
Carcinoma neuroendocrino de grado 3:. tumor mal diferenciado de célula pequeña o célula grande, índice Ki-67 mayor de 20 % e índice mitótico mayor de 20 por 10 campos de mayor aumento
Rendimiento del índice Ki-67 en tumores neuroendocrinos de páncreas: el índice Ki-67 es un requisito para la clasificación y, junto con el conteo mitótico, son los factores pronósticos más fiables en la actualidad para los tumores neuroendocrinos de páncreas. Se basa en el conteo de, al menos, 500 células en las áreas de mayor proliferación o “puntos calientes” (hot spots); sin embargo, el método para su cuantificación sigue siendo materia de gran debate. En la actualidad, se recomienda hacer un conteo manual con imágenes impresas, evitando hacerlo a simple vista. Este método es el más sencillo, reproducible, preciso y económico 19. En la figura 1 se muestra la patología de un tumor neuroendocrino de páncreas de grado 1 dado por el grado de diferenciación e índice Ki-67.
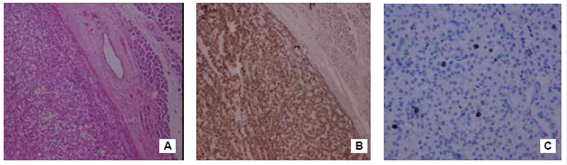
Figura 1. Tumor neuroendocrino de páncreas bien diferenciado de grado 1. A) En el lado izquierdo se aprecia el tumor y, al lado derecho, el tejido pancreático normal. Hematoxilina y eosina, 10X. B) Tinción de inmunohistoquímica positiva para cromogranina, 10X, C) Tinción de inmunohistoquímica para el índice de proliferación Ki-67 que es menor de 2 %, 10X.
El índice de proliferación celular Ki-67 mayor de 55 % predice una mejor respuesta a la quimioterapia basada en cisplatino, mientras que, con uno menor de 55%, hay mayor supervivencia con pobre respuesta a la quimioterapia 13,14.
Estadificación TNM: se ha propuesto la modificación de este sistema de evaluación de carga tumoral, basándose en las curvas de supervivencia y en la importancia de subclasificar la estadificación ganglionar. En la última clasificación de 2016 propuesta por la European Neuroendocrine Tumor Society (ENETS), se modifican los grupos pronósticos de estadio tumoral y se adoptan los propuestos por la American Joint Committee on Cancer (AJCC), pero se mantienen los criterios para determinar el estadio del tumor primario, de los ganglios linfáticos y de las metástasis 20.
Evaluación clínica
La anamnesis es clave; la historia personal y familiar es muy importante para determinar si se trata de un tumor neuroendocrino esporádico o heredofamiliar. Los sujetos con historia personal de dos o más tumores neuroendocrinos o una historia familiar de tumor neuroendocrino, deben ser evaluados para el síndrome genético correspondiente. Es necesario valorar los síntomas producidos por la secreción hormonal o por el efecto de masa tumoral, para determinar la funcionalidad tumoral; en particular, se debe interrogar sobre la presencia de lipomas, lesiones en la piel, y antecedentes de urolitiasis y otros tumores no endocrinos. Se debe evaluar el estado funcional usando el índice de Karnosfky (IK) o la escala del Eastern Cooperative Oncology Group (ECOG).
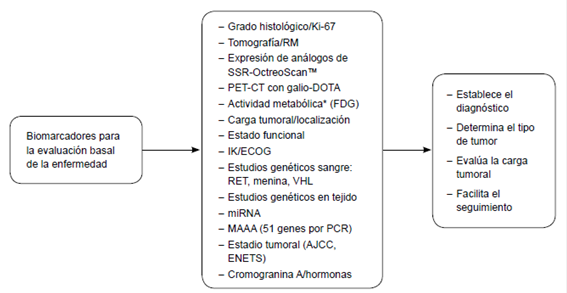
RM: ResonanciaMagnética; SSR: Receptor de Somatostatina; IK: Indice de Karnofsky; ECOG: Escala Eastern; Cooperative Oncology Group; MAAA: multianalyte algorithmic assay; AJCC: American Joint Committee on Cancer; ENETS: European Neuroendocrine Tumor Society. * PET CT con 18 FDG
Figura 2. Biomarcadores que pueden ser útiles en la evaluación de tumores neuroendocrinos de páncreas.
La presentación clínica define si el tumor es funcional o si no lo es; sin embargo, puede ser un reto diagnóstico. Hasta 60 a 70 % de los casos se presentan con metástasis, en su mayoría hepáticas 4.
A continuación, se describen brevemente los tumores neuroendocrinos funcionales. En la tabla 1 se resumen los síndromes producidos por los tumores neuroendocrinos del páncreas.
Tabla 1. Síndromes producidos por los tumores neuroendocrinos de páncreas. La frecuencia reportada está en relación con los tumores neuroendocrinos de páncreas. Es necesario recordar que estos tumores, en fases avanzadas, pueden volverse funcionales y multisecretores (secreción metacrónica).
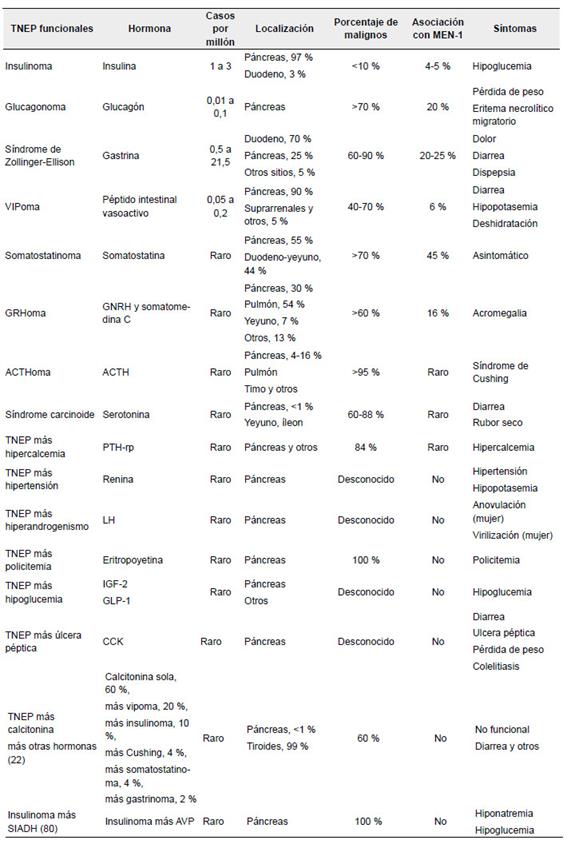
TNEP: tumores neuroendocrinos del páncreas: VIP: Vasoactive Intestinal Peptide; ACTH: Adrenocorticotropic hormone; GnRH: Gonadotropin-releasing hormone; PTH-rP: Parathyroid hormone-related protein; LH: Luteinizing hormone; IGF-2: Insulin-like growth factor 2; GLP-1: Glucagon-like peptide-1; CCK: Cholecystokinin; SIADH: Syndrome of Inappropriate Antidiuretic Hormone Secretion; AVP: Arginine vasopressin.
Insulinoma
Se deriva de las células beta de los islotes de Langerhans y es más frecuente en mujeres, con una relación 2:1 frente a los hombres en la quinta a sexta década de la vida. Secreta insulina de manera autónoma, lo que conlleva hipoglucemia.
La presentación clínica se caracteriza por la tríada de Whipple (hipoglucemia, síntomas neurológicos y recuperación con la ingestión de glucosa). Estos síntomas se deben al efecto de la hipoglucemia sobre el sistema nervioso central; la mayoría de los pacientes tiene síntomas secundarios a un exceso de liberación de catecolaminas por la hipoglucemia, lo que incluye sudoración, temblor y palpitaciones. Menos del 10 % están asociados con el síndrome MEN-1 (neoplasia endocrina múltiple de tipo 1) y suelen presentarse con focos múltiples. El 95 % son pequeños, miden menos de 2 cm, y son malignos solamente en 5 a 10 % de casos 4,21,22.
Gastrinoma
Es el segundo tumor neuroendocrino funcional de páncreas y se localiza en el triángulo del gastrinoma, formado por la unión del conducto cístico y el colédoco, la segunda y la tercera porciones del duodeno y el cuello del páncreas. El 70 % de estos tumores se ubican en el duodeno, el 25 % en el páncreas, y el 5 % en tejidos adyacentes. La edad media del diagnóstico es 50 años, es más frecuente en hombres y se asocia con alcoholismo.
A pesar de tener un crecimiento lento, hasta del 60 al 90 % tienen un comportamiento maligno, con diseminación metastásica a los ganglios linfáticos y al hígado en un tercio de los casos en el momento del diagnóstico.
Es un tumor secretor de gastrina que lleva al síndrome de Zollinger-Ellison. Este consiste en úlceras pépticas que suelen ser resistentes al tratamiento, asociadas con dolor abdominal, diarrea, pirosis, náuseas y pérdida de peso. Las úlceras pueden presentarse en sitios poco comunes, sin embargo, es característica la úlcera duodenal típica. Alrededor del 30 % de los pacientes tiene síndrome MEN-1 4,21,23.
Glucagonoma
Es un tumor que secreta cantidades excesivas de glucagón, diagnosticado en la quinta década de la vida, y con igual prevalencia entre hombre y mujeres.
Todos los glucagonomas aparecen en el páncreas, principalmente en la cola. Usualmente, son tumores grandes (>5 cm), malignos en más del 60 % de los casos, con metástasis a hígado en hasta en el 80 %. Del 5 al 17 % se asocia con el síndrome MEN-1. Este se caracteriza por pérdida de peso, queilitis, dermatitis (eritema necrolítico migratorio), diabetes mellitus, diarrea, estreñimiento, dolor abdominal, anorexia, tromboembolia venosa profunda y síntomas neurológicos, como ataxia, depresión y debilidad muscular proximal 7,21,23.
Vipomas
Son tumores raros, tienen una incidencia de 1 por un millón por año, y suelen ser esporádicos y grandes, de más de 3 cm. El 95 % se presenta en el páncreas, principalmente en la cola, pero pueden localizarse en bronquios, colon, glándulas suprarrenales e hígado. El 90 % son malignos y 60 a 80 % presentan metástasis al momento del diagnóstico.
Secretan gran cantidad de péptido intestinal vasoactivo (VIP), lo que origina el síndrome de Verner-Morrison, caracterizado por diarrea líquida de gran volumen (cólera pancreática), hipocalcemia y aclorhidria. Otros síntomas que se pueden encontrar son: rubor o rubefacción, dolor abdominal, insuficiencia renal, hipercalcemia, hiperglucemia, náuseas, calambres, debilidad y deshidratación 7,21,23.
Somatostatinoma
Es un tumor raro que se presenta en menos de 1 de 40 millones de personas. El 50 % ocurre en el páncreas, y el 50 % en duodeno y yeyuno. Es maligno en 6 a 70 % de los casos y las metástasis hepáticas son frecuentes (80 %). Se pueden asociar con el síndrome MEN-1 o el MEN-2, la neurofibromatosis (gen NF1) y la enfermedad de von Hippel-Lindau (gen VHL). El 98 % de los somatostatinomas intestinales son asintomáticos; se ha cuestionado la existencia de un síndrome clínico 7,21,23,24.
GRFoma
Son tumores muy raros y no se conoce su incidencia. Son malignos en 30 a 50 % de los casos y puede originarse en el páncreas, el pulmón y el intestino delgado. Secretan hormona del crecimiento y causan acromegalia.
ACTHoma
Es un tumor maligno el 95 % de las veces, más frecuente en mujeres que en hombres. Secreta ACTH y es responsable del 4 % de los casos de síndrome de Cushing. Puede asociarse con el síndrome de Zollinger-Ellison hasta en 8 % de los pacientes, y del 26 al 80 % de estos tumores secretan gastrina.
PTHrp-oma
Son tumores raros y, a pesar de ser de bajo grado, más del 85 % son malignos. Causan hipercalcemia por producción de PTHrp (péptido relacionado con la parathormona).
Tumores neuroendocrinos de páncreas que causan síndrome carcinoide
Existen menos de 50 casos reportados en la literatura científica y se ubican principalmente en la cola del páncreas; entre el 60 y el 90 % de estos tumores son malignos. Corresponden a menos del 1 % de los carcinoides gastrointestinales y son de mejor pronóstico. Estos tumores secretan serotonina y taquicininas, lo cual causa rubor o rubefacción, diarrea y dolor abdominal.
Evaluación bioquímica y hormonal
Los biomarcadores consisten en expresiones celulares, bioquímicas o moleculares que se pueden rastrear o medir mediante herramientas tecnológicas que facilitan la predicción, aclarar la etiología, hacer el diagnóstico, y valorar la evolución (progresión o regresión) de la enfermedad y el resultado del tratamiento 25.
Los biomarcadores permiten integrar la génesis tumoral y las manifestaciones clínicas, lo cual repercute en la estrategia terapéutica; sin embargo, en forma aislada, no permiten hacer el diagnóstico. Entre sus ventajas están: se correlacionan con la carga tumoral, conducen al diagnóstico temprano, evalúan la enfermedad residual mínima, apoyan la elección de determinada modalidad diagnóstica compleja, y pueden demostrar el éxito o el fracaso del tratamiento 26.
Entre sus desventajas, se incluyen: no estar disponible en todos los centros, los altos costos, su difícil uso en enfermedades avanzadas, y la variabilidad en la sensibilidad y la especificidad.
Debido a la complejidad, la heterogeneidad y el fenotipo de los tumores neuroendocrinos de páncreas, no existe un único biomarcador para ellos; son múltiples y se pueden evaluar en tejidos, sangre y orina, así como con imágenes combinadas con radionúclidos, con gran variabilidad en la sensibilidad, la especificidad y la reproducibilidad debido a cambios en la metodología y la estandarización de las técnicas.
Biomarcadores generales
Cromogranina A. Es un marcador ampliamente usado en la última década en tejido y en sangre; se expresa en los sujetos sanos, en enfermedad neoplásica y no neoplásica. Su rendimiento es superior cuando se ha confirmado el tumor neuroendocrino y es ineficaz como marcador diagnóstico aislado. Se correlaciona con la carga tumoral, con la enfermedad metastásica y con el pronóstico. No se correlaciona con la funcionalidad ni la localización tumoral. Las muestras sanguíneas se deben tomar en ayunas con suspensión previa de los inhibidores de bomba de protones, por lo menos, dos semanas antes 27. Hay gran variabilidad entre los ensayos comerciales para la cromogranina A 28.
Pancreastatina. Es el polipéptido más corto de la cromogranina A y se encuentra elevada en 58 a 81 % de los tumores neuroendocrinos; se puede medir en sangre y orina 29. No se correlaciona con la carga tumoral, la localización, la agresividad ni la supervivencia. Se afecta por la ingestión de alimentos o la hiperglucemia 30.
Cromogranina B y C. El rendimiento de las cromogranina B y C en tumores neuroendocrinos es inferior al de la cromogranina A. Al parecer, no se afectan por el deterioro de la función renal o el uso de inhibidores de bomba de protones 31.
Enolasa neuronal específica. Está presente en neuronas y células neuroendocrinas. Hasta el momento, no tiene una buena utilidad clínica 32.
Biomarcadores específicos
Polipéptido pancreático. Se produce en las células neuroendocrinas del páncreas y del colon, desempeñando un papel en la autorregulación de la secreción. Su utilidad clínica es limitada 33,34.
Gastrina. Es un marcador que se puede evaluar en sangre y tejido. En caso de gastrinoma, la gastrina sérica en ayunas es mayor de 100 pg/ml o más de 10 veces el límite superior, el pH gástrico es menor de 2, la salida de ácido basal es mayor de 15 mEq/L y la prueba de secretina muestra un aumento de la secreción de gastrina.
Es importante recordar que la gastrinemia es alta en la atrofia gástrica y que los inhibidores de la bomba de protones se deben suspender, al menos, una semana antes de la toma de exámenes y, los antagonistas H2, dos días antes 4,21,35.
Insulina y prueba en ayunas. El diagnóstico y el seguimiento de los insulinomas requieren confirmar: valor de glucosa inferior a 40 mg/dl, insulina mayor de 36 pmol/L, péptido C mayor de 200 pmol/L, proinsulina de más de 5 pmol/L y β-hidroxibutirato inferior a 2,7 mmol/L. Además, se debe documentar la ausencia de sulfonilureas o sus metabolitos en plasma u orina y una buena respuesta terapéutica con el glucagón 21.
Glucagón. Es un marcador que se puede evaluar en sangre y tejido. El síndrome de glucagonoma conlleva valores de glucagón plasmático por encima de 500 pg/ml y elevación de la glucosa en sangre 7,21.
Péptido intestinal vasoactivo. Este marcador se evalúa en sangre. Los niveles encontrados en caso de VIPoma son mayores de 500 pg/ml y, en la diarrea secretoria aguda, mayores de 700 ml/d 21.
Somatostatina. Es un marcador que se evalúa en sangre y tejido. Para el diagnóstico del somatostatinoma, se requiere elevación de la somastotatina sérica y la inmunohistoquímica positiva en el tejido.
Otros biomarcadores menos frecuentes y su síndrome
GRFoma. Causa aumento de la hormona del crecimiento, elevación de la somatomedina C y hábito acromegálico. La prueba de supresión de glucosa para la hormona del crecimiento es positiva, con imágenes que descartan tumores hipofisiarios.
ACTHoma. Produce elevación de la ACTH sérica, del cortisol libre en orina de 24 horas y del cortisol salival de las 11 p.m. La prueba de supresión con dexametasona es positiva y existe hábito cushingoide con coloración parda de la piel. Se deben descartar tumores hipofisiarios y, por medio del cateterismo de los senos petrosos, confirmar la producción extrahipofisiaria.
PTHrp-oma. Produce hipercalcemia, elevación de la PTH-rp y supresión de la PTH endógena.
Tumor neuroendocrino de páncreas que causa síndrome carcinoide asociado a elevación de serotonina y sus metabolitos. Se puede medir el ácido 5-hidroxiindolacético en orina de 24 horas.
Biomarcadores emergentes
Células tumorales circulantes. La detección de células tumorales circulantes se basa en la tecnología que captura la expresión celular de las moléculas de adhesión celular epitelial en varios tumores sólidos. En los tumores neuroendocrinos metastásicos se encuentran células tumorales circulantes detectables en el intestino medio (43 %) y el páncreas (21 %), sin correlación con el índice Ki-67 y la cromogranina A sérica 36. Alrededor del 50 % de los pacientes con tumores neuroendocrinos tienen células tumorales circulantes detectables y se correlacionan con el compromiso hepático 37.
MicroARN. Son ARN no codificantes de pequeño tamaño (19 a 25 nucleótidos) que regulan los mecanismos posteriores a la transcripción en diversos procesos; se encuentran alterados en neoplasias y son potenciales biomarcadores y agentes terapéuticos. Se pueden evaluar en sangre y en tejido, pero hay dificultades para su estandarización y reproducibilidad.
En los tumores neuroendocrinos de páncreas se ha encontrado aumento de la regulación de miR-2, miR-103, miR107, miR-193b. La sobreexpresión de miR-21 se asocia con un alto índice Ki-67 y metástasis hepáticas. La expresión de miR-642 se correlaciona con el índice Ki-67 y la expresión de miR-210 se asocia con enfermedad metastásica. Se ha encontrado disminución de la regulación de miR-584, miR-1285, miR-550-002410, miR-1290 y miR-1825; la disminución de la regulación sérica del miR-1290 discrimina los tumores neuroendocrinos de páncreas de los adenocarcinomas 38,39,40.
Análisis genético por transcripción por PCR, microarreglos. La tasa de mutaciones de los tumores neuroendocrinos de páncreas es de 0,82 por megabase (rango de 0,04 a 4.56) y es baja al compararse con la del adenocarcinoma de páncreas, que reporta 2,64 (rango de 0,65 a 28,2). Múltiples genes candidatos asociados se pueden medir en sangre o tejido fresco. Las pruebas de PCR son estandarizadas y reproducibles, con un coeficiente de variabilidad entre ensayos e intraensayos inferior al 2 %; y no se afectan por edad, sexo, etnia, ayuno o medicación 41,42.
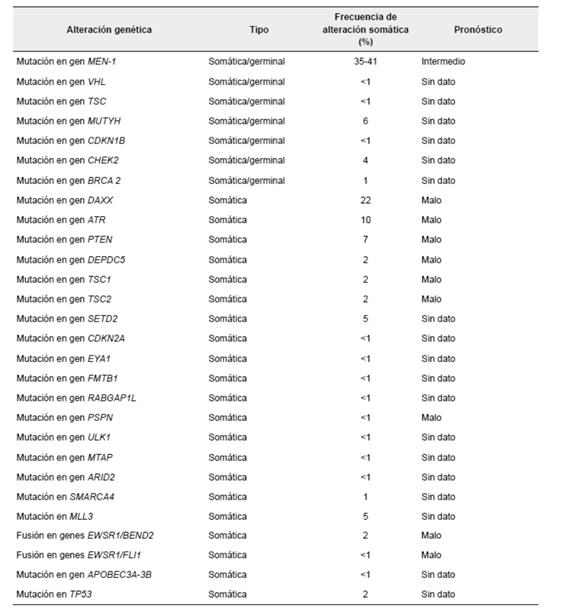
En los tumores neuroendocrinos de páncreas bien diferenciados, se pueden encontrar genes con mutaciones somáticas múltiples, como: MEN1, DAXX, ATRX, TSC, NF1 y MUTYH (tabla 2) 43.
Tabla 2. Alteraciones genéticas halladas en tumores neuroendocrinos de páncreas, estudios realizados en pequeño número de poblaciones.
NETest. Permite evaluar, en sangre periférica, la expresión de ARN de 51 genes para identificar tumores neuroendocrinos gastroenteropancréaticos. Ha demostrado buen rendimiento en pacientes con tumores bien y moderadamente diferenciados de intestino medio y páncreas metastásicos 41,42. Hasta marzo de 2018, se había evaluado en 5.207 pacientes de 106 instituciones de Estados Unidos y Europa con una probabilidad diagnóstica mayor del 90 %, un valor predictivo positivo mayor del 92 % y valor predictivo pronóstico mayor del 95 %; se ha evaluado en pretratamiento para la predicción de la respuesta a la terapia con radionúclidos con una exactitud del 93 % 44.
Evaluación radiológica
La evaluación de la estadificación tumoral inicial y el seguimiento recae principalmente en las imágenes diagnósticas como la tomografía computadorizada (TC) y la resonancia magnética (RM). Las técnicas de medicina nuclear combinadas (imágenes híbridas) con TC y RM mejoran la localización de los tumores ocultos y mejoran la estadificación, como la SPECT (TC de emisión monofotónica), la gammagrafía con análogos del receptor de somatostatina (RSST), la tomografía con emisión de positrones (PET) con análogos del RSST y, en algunas ocasiones, la PET con 18F-DOPA o con 18F-FDG. Las innovaciones en medicina nuclear con antagonistas para el RSST y agonistas del receptor GLP-1 (Glucagon-Like Peptide-1), podrían mejorar las imágenes de los tumores neuroendocrinos de páncreas 45.
Radiología convencional
Ultrasonografía de abdomen. Desempeña un papel secundario en el seguimiento; se utiliza en el seguimiento temprano de los tratamientos con radionúclidos, en radiofrecuencia, formación de émbolos y quimioémbolos de metástasis hepáticas.
Tomografía computarizada. Es una de las imágenes diagnósticas de elección para el diagnóstico y el seguimiento de los tumores neuroendocrinos de páncreas. Se requiere un protocolo con contraste multifásico y, como mínimo, debe tener una fase sin contraste, una fase venosa portal y una fase arterial. Estos tumores, generalmente, presentan una hipercaptación en la fase arterial; sin embargo, algunos tumores no la presentan o lo hacen en la fase tardía. La TC es la imagen de elección para el seguimiento, porque se ha valorado objetivamente su uso mediante los criterios RECIST, RECIST modificado, Chun y Choi.
Resonancia magnética. Es otra de las imágenes diagnósticas de elección para el diagnóstico y el seguimiento. Se requiere un protocolo con contraste dinámico o multifásico para mejorar la sensibilidad que reporte imágenes morfológicas neutras (ponderadas) en T1 y T2, e imágenes con saturación de grasa en T2. Se deben hacer la supresión de grasa y la compensación de movimiento, y se debe incluir la difusión de imágenes ponderadas para la parte superior del abdomen.
En los tumores neuroendocrinos de páncreas con metástasis hepáticas, la sensibilidad es mejor en la fase de difusión pesada y con el uso de medios de contrastes específicos para el hígado. Algunas características pueden sugerir mayor agresividad tumoral, como: lesiones tumorales de gran diámetro, signos de invasión vascular, isointensidad o hipointensidad en la fase venosa portal, ausencia de plano de clivaje, compresión del conducto biliar y extensión extrahepática 46,47.
En las imágenes de difusión pesada, los valores bajos del coeficiente de difusión aparente (Apparent Diffusion Coefficient, ADC) se asocian con mayor densidad celular, abundancia de organelos celulares y mayor agresividad tumoral 47,48.
Las ventajas de la RM es que no produce radiación ionizante, tiene mayor sensibilidad para la detección de metástasis hepáticas y ofrece otras técnicas funcionales.
Imágenes de medicina nuclear
Las imágenes de medicina nuclear son utilizadas para la estadificación, el seguimiento y la reestadificación, ya que los tumores neuroendocrinos de páncreas poseen características moleculares relevantes que permiten su uso, como: expresan RSST en 80 a 100 % de casos, especialmente el subtipo 2 (RSST2) 49, pero, también, expresan receptores para GLP-1 y GIP (péptido insulinotrópico) 50, tienen capacidad para captar y decarboxilar aminas 51 y, en general, bajo consumo de glucosa. La mayoría expresan gran cantidad de RSST2, a excepción de los insulinomas, por lo que constituyen un blanco atractivo para imágenes y tratamiento con radionúclidos. En la tabla 3 se presenta el rendimiento diagnóstico de las diferentes imágenes diagnosticas en los tumores neuroendocrinos de páncreas. En Colombia, tenemos algunas modalidades como la gammagrafía con análogos del RSST y OctreoScan™, PET/CT con galio 68 y PET/CT con 18 FDG.
Tabla 3. Rendimiento de las imágenes diagnósticas en tumores neuroendocrinos de páncreas, con utilidad tanto para estadificación como para hacer seguimiento y evaluar la respuesta.
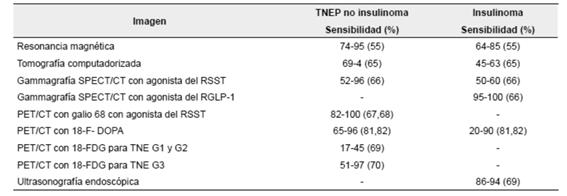
TNEP: tumores neuroendocrinos de páncreas; TNE: tumores neuroendocrinos
Gammagrafía con análogos del RSST y OctreoScan™ con SPECT/CT. Su blanco es el RSST2, útil en tumores neuroendocrinos de páncreas bien y moderamente diferenciados. El 111In-ácido dietilenetriaminopentaacético-D-Fe1-octreótido (111In-DTPA-octreótido), conocido como OctreoScan™, sigue siendo la modalidad diagnóstica estándar, aunque se utilizan alternativas más económicas, más flexibles, validadas y con tiempos más tempranos de adquisición de imágenes, como: el 99mTc-EDDA-hidrazinonicotinil-Tir3-octreótido (99mTc-HYNIC-TOC) y el 99mTc-EDDA-hidrazinonicotinil-Tir3-octreotato 52.
Todas tienen limitaciones similares, como baja resolución espacial, baja sensibilidad en la detección de pequeños tumores, bajo contraste con órganos normales como hígado, riñón y bazo, y bajo rendimiento en insulinomas. Puede presentar falsos positivos en lesiones inflamatorias y en otros tumores no endocrinos 53.
Tomografía por emisión de positrones (PET/CT) con análogos del RSST. Las imágenes con agonistas del RSST marcadas con galio-68 tienen varias ventajas sobre el OctreoScan™ como: adquisición de imágenes de alta calidad a los 45 minutos de inyectar el radiotrazador, mayor resolución espacial, cuantificación de la captación del radiotrazador expresadas en SUV (Standard Uptake Value) y menor dosis de radiación. Es la imagen de elección actual, porque cambia la estrategia de manejo en 70 % de los casos 54. Se han encontrado falsos negativos en lesiones menores de 7 mm, en tumores neuroendocrinos pobremente diferenciados y en insulinomas; también, falsos positivos principalmente en procesos inflamatorios 55 y lesiones intracerebrales, como meningiomas.
Se han marcado diferentes péptidos con galio-68 y han mostrado diferente afinidad por el subtipo de RSST, pero en la práctica tienen igual eficiencia, entre ellos están 55,56,57:
- [68Ga-DOTA,Tir3]-octreótido: 68Ga-DOTATOC,
- [68Ga-DOTA,Tir3]-octreotato: 68Ga-DOTATATE que tiene mayor afinidad por el RSST2, pero no se liga con otros subtipos, y
- [Ga68-DOTA,1-Nal3]-octreótido: 68Ga-DOTANOC: tiene mayor contraste con la actividad hepática normal.
PET/CT con 18F-FDG (fluorina-18-fluorodeoxiglucosa). Evalúa el consumo de glucosa por los diferentes tejidos y cuantifica la captación en SUV (Standard Uptake Value); sirve para caracterizar las formas más agresivas de los tumores neuroendocrinos de páncreas como los de grado 3, los pobremente diferenciados o ambos 59. Un reporte negativo se correlaciona con un mejor pronóstico y una mayor tasa de supervivencia 50. Un reporte positivo es predictivo de gran agresividad y baja tasa de supervivencia, incluyendo un tumor neuroendocrino con un índice Ki-67 menor de 2 % 60,61.
PET con 18F-DOPA (fluorina-18-dihidroxifenilalanina). El 18F-DOPA es un precursor de la captación de la decarboxilación de aminas (APUD) y es captado por los tumores neuroendocrinos mediante el transportador LAT2. Tiene gran sensibilidad en el estudio del hiperinsulinismo congénito, y puede ser útil en el estudio de insulinomas, paragangliomas, feocromocitomas y tumores secretores de serotonina. En los tumores neuroendocrinos de páncreas, tiene mayor rendimiento comparado con el OctreoScan™, pero es inferior al PET/CT con galio-68 61,62,63.
Gammagrafía con 123I-metayodobenzilguanidina con SPECT/CT. La metayodobenzilguanidina (MIBG) es un análogo de noradrenalina que se marca con 123I y cruza la membrana plasmática mediante el transportador de norepinefrina humano, acumulándose en los tumores neuroectodérmicos. Tiene baja sensibilidad en los tumores neuroendocrinos de páncreas, por lo tanto, no se recomienda de rutina 55.
Otras modalidades de evaluación
Gammagrafía con agonistas para el receptor GLP-1 con SPECT/CT. El receptor de GLP-1 es expresado con alta incidencia y densidad en los insulinomas benignos, simplificando el diagnóstico y el proceso quirúrgico 64,65,66. Entre las modalidades, tenemos:
- [Lis40(Ahx-DOTA-111In)NH2] exendin-4: 111In-DOTA-exendin-4,
- [Lis40(Ahx-DTPA-111In)NH2] exendin-4: 111In-DTPA-exendin-4 y
- [Lis40(Ahx-HYNIC-99mTc/EDDA)NH2] exendin-4: 99mTc-HYNIC-exendin-4.
PET/CT con agonistas para el receptor GLP-1. Estas técnicas tienen mayor resolución espacial, sensibilidad y exactitud, y son más efectivas en la detección de insulinomas comparadas con la gammagrafía con SPECT/CT y las imágenes morfológicas 58,59. Entre las modalidades, tenemos:
- [68Ga-DO3A-VS,Cis40]exendin-4,
- [Nle14, Lis40(Ahx-DOTA-68Ga)NH2]exendin-4: 68Ga-DOTA-exendin-4 y
- [Lis40(Ahx-NODAGA-68Ga)NH2]exendin-4: 68Ga-NODAGA-exendin-4.
Gammagrafía con 64Cu-DOTATATE. Por las dificultades en la adquisición del PET/CT con galio 68, se ha evaluado el cobre radiactivo, el cual ha mostrado superioridad sobre el OctreoScan™ 60.
Gammagrafías y PET/CT con antagonistas del RSST. Esta modalidad ha demostrado mayor captación, mayor tiempo de retención y más sitios de unión del radiotrazador en el tumor sin interiorización, y ha demostrado tener mayor sensibilidad; entre ellos, tenemos:
Gammagrafía con agonistas del receptor GIP. El receptor GIP se expresa en los tumores neuroendocrinos bien y moderadamente diferenciados, pero no hay estudios al respecto.
Evaluaciones invasivas
Ultrasonografía endoscópica. Se usa cuando las imágenes por RM y TC no han localizado el tumor 73. Tiene la ventaja de que se pueden tomar muestras histológicas mediante biopsia con aguja fina, con mínima invasión y disminución de los eventos adversos.
Estímulo selectivo de calcio arterial y cateterismo venoso hepático. Son modalidades para detectar y localizar tumores pequeños con mayor sensibilidad, generalmente, en la evaluación del insulinoma y el gastrinoma no localizados con los estudios convencionales 74.
Evaluación genética
Si el paciente presenta dos o más tumores neuroendocrinos, o estos se han detectado en dos o más miembros de la familia, se requiere el estudio de enfermedades heredofamiliares que, dependiendo del fenotipo expresado, hará la evaluación más costo-efectiva.
El síndrome más frecuente que involucra tumores neuroendocrinos de páncreas es el MEN-1. Este está presente en 20 a 25 % de los pacientes con gastrinomas, en 4 % de aquellos con insulinomas y en menos de 3 % en otros tumores neuroendocrinos de páncreas. Casi todos los pacientes con MEN-1 tienen múltiples focos de tumores no funcionales y asintomáticos. Otros, son el síndrome MEN-4 (gen CDKN1) 75,76y la enfermedad de von Hippel-Lindau (gen VHL) 77. Recientemente, se han caracterizado la deficiencia en el gen MUTYH que se asocia con predisposición a cáncer de colon, cáncer gástrico y otros no gastrointestinales 78, y la mutación de CHEK2, que se asocia con carcinoma de mama y de próstata 79.
Conclusiones
En la actualidad, se cuenta con múltiples biomarcadores para el abordaje inicial y el seguimiento de los tumores neuroendocrinos, y en los gastroenteropancréaticos se tiene la mayor evidencia al respecto. No existe un biomarcador perfecto debido a la complejidad y la heterogeneidad de los tumores neuroendocrinos. Dependemos de la evaluación clínica, radiológica y de la experiencia médica asociada al recurso disponible.
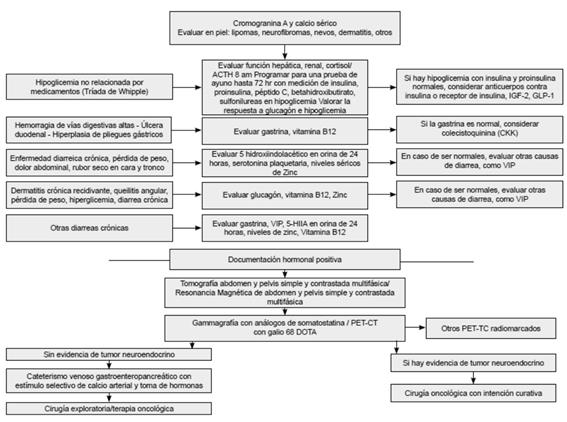
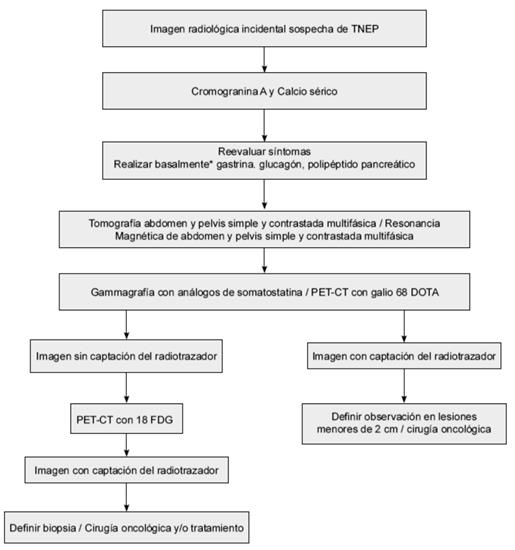
Paciente con sospecha de tumor neuroendocrino pancreático funcional
Figura 3. Algoritmo diagnóstico en tumores neuroendocrinos funcionales de páncreas.
Los biomarcadores séricos y urinarios son difíciles de medir y tienen importante variabilidad preanalítica y analítica. Sin embargo, en los tumores neuroendocrinos funcionales, los biomarcadores séricos tienen mayor rendimiento y son necesarios para establecer el diagnóstico. Un solo estudio, por sí mismo, no brinda la suficiente información diagnóstica, predictiva ni pronóstica. La cromogranina A está sobrevalorada en la práctica clínica y es necesario estandarizarla para nuestra población y conocer muy bien los falsos positivos.
Disponemos de múltiples estudios genéticos, sin embargo, muchos estudios de mutaciones genéticas en tejido y en sangre, no tienen estudios de validación pronóstica ni confirmatoria en los tumores neuroendocrinos de páncreas. Los miRNA han sido evaluados en forma exploratoria y no hay estudios confirmatorios. Las alteraciones epigenéticas han sido evaluadas en pequeños estudios y les falta validación prospectiva. La evaluación multianalítica tiene el potencial de mayor sensibilidad y especificidad en la evaluación inicial y seguimiento, pero requiere revalidación en múltiples circunstancias. Por ahora, se deben practicar estudios genéticos si hay sospecha de síndrome herodofamiliar, sea por el antecedente familiar o por la sintomatología del paciente.
En resumen, una buena historia clínica aunada al conocimiento de la patología, con el uso rutinario del índice Ki-67 y del conteo mitótico, nos ayuda a definir las mejores herramientas para el abordaje del paciente con tumores neuroendocrinos de páncreas y planear el mejor tratamiento y seguimiento.