











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La primera descripción de las glándulas paratiroideas como órgano fue hecha en 1850 en el zoológico de Londres durante una necropsia a un rinoceronte. La primera cirugía de paratiroides fue realizada en 1925 en Viena por Félix Mandl en un paciente con una osteítis fibrosa quística. En ese mismo año, el bioquímico James B Collip, en Canadá, descubrió la hormona paratiroidea, (PTH, por sus siglas en inglés) después de participar en el grupo de investigadores que aislaron la insulina y la hormona adrenocorticotrópica (ACTH).
El hiperparatiroidismo primario (HPTP), es el tercer trastorno endocrino en frecuencia 1, que se caracteriza porque, en ausencia de un estímulo reconocido, una o varias glándulas paratiroideas secretan en exceso PTH, resultando en hipercalcemia. Se estima que su prevalencia es de 0,85 % y su incidencia actual es de aproximadamente 50 casos por cada 100.000 personas-año en Estados Unidos, siendo más frecuente en mujeres (2 a 3:1) entre 50-65 años de edad 2.
La presentación clínica ha variado en los últimos 40 años, desde cuadros muy sintomáticos asociados a hipercalcemia severa, litiasis renal y enfermedad ósea (osteítis fibrosa quística), hasta una condición asintomática, más frecuente en la actualidad, diagnosticada de forma incidental con exámenes rutinarios (calcio sérico, pruebas bioquímicas, densitometría ósea, etc.)
Embriología y anatomía
Las glándulas paratiroideas se desarrollan entre la quinta y séptima semana de gestación. Las glándulas superiores se originan de la cuarta bolsa branquial y generalmente se localizan hacia la unión cricotraqueal, entre el tejido tiroideo y la rodilla del nervio laríngeo recurrente, mientras las glándulas inferiores se originan de la tercera bolsa branquial. Aproximadamente el 44 % se encuentran en el ligamento tirotímico, el 26 % en el cuerno tímico, el 17 % en el polo inferior de la glándula tiroidea, 2 % en el mediastino y el 11 % están distribuidas en el lecho carotídeo superior o dentro del tiroides 3.
En el 91 % de las personas son 4 glándulas, en el 5 % son 3 y en el 4 % son 5 o más. Habitualmente cada una mide entre 5 y 7 mm. Todo el tejido paratiroideo puede pesar entre 90 y 130 mg. En el 83 % de los casos tienen forma ovalada, en 11 % elongada, 5 % arriñonada y 1 % multilobulada. Generalmente se acompañan de tejido graso periglandular y en el 80 % de los casos su ubicación contralateral es en espejo 3. Usualmente las paratiroides reciben su irrigación de la arteria tiroidea inferior, sin embargo, hasta en un 45 % de los pacientes, las glándulas superiores pueden recibir ramas de la arteria tiroidea superior 4.
Homeostasis del calcio
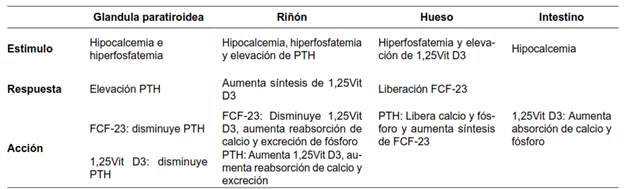
La ingesta diaria de calcio en promedio es de 500 a 1000 mg. Su absorción ocurre principalmente en el duodeno y yeyuno proximal. En el riñón, casi todo el calcio filtrado se reabsorbe a nivel del túbulo proximal y el asa de Henle, a través de la bomba de sodio. En el túbulo distal, la absorción del calcio está mediada por la PTH. Las pérdidas diarias de calcio son aproximadamente de 100 mg por transpiración y 800 mg en las heces. El mayor depósito corporal de calcio está en los huesos, que contiene aproximadamente 1000 gr 5.
El control de los niveles séricos de calcio y fósforo es complejo, porque no sólo depende de la ingesta, sino de la participación de órganos como intestino (donde ocurre la absorción mediada por metabolitos de la vitamina D), riñón (excreción), hígado (fragmentación de la PTH), hueso (depósito y liberación), piel (activación de la vitamina D3 por irradiación ultravioleta del 7-dihidroxicolesterol), glándulas paratiroideas (hormona paratiroidea) y glándula tiroides (donde las células C o parafoliculares producen Calcitonina, la cual disminuye la reabsorción ósea por su efecto antagónico a la PTH).
Las concentraciones de calcio sérico normalmente deben mantenerse en un rango muy estrecho y preciso, entre 8,5-10,5 mg/dl o 2,1-2,6 mmol/l, que es el valor que se requiere para la realización óptima de múltiples procesos fisiológicos, intra y extracelulares en el organismo. Lograr la regulación de su concentración con exactitud, depende de la relación entre la absorción intestinal, la excreción renal y la captación y liberación ósea, procesos que a su vez están regulados por la acción de la hormona paratiroidea, la vitamina D3 y, en menor participación, de la calcitonina.
La PTH es la principal hormona reguladora de la homeostasis del calcio. Es producida por las células principales de la glándula paratiroides, en forma de una prohormona de 110 aminoácidos, que se divide y se convierte finalmente en la hormona propiamente dicha, un polipéptido de 84 aminoácidos. Tiene una porción biológicamente activa que corresponde al grupo amino N-terminal (residuos 1-34) y una inactiva que corresponde al grupo carboxilo C-terminal (residuos 35 a 84). La vida media de la PTH intacta es de aproximadamente 6 minutos en el fragmento N-terminal y casi 1 hora en el fragmento C-terminal. Su degradación ocurre principalmente en el hígado.
El calcio constituye el principal mineral corporal, pero solamente su forma ionizada es fisiológicamente activa. En suero se presenta 47 % en forma ionizada, 45 % ligado a proteínas y 8 % en forma de aniones orgánicos. Su función es estimular la contracción muscular, la excitabilidad nerviosa, la formación de matriz ósea y facilitar la transducción de señales 5.
La hipocalcemia es el principal estímulo fisiológico de las células principales en las glándulas paratiroideas, donde mediante tres receptores se regula la síntesis y secreción de la hormona paratiroidea:
El receptor de la vitamina D (RVD) que, al unirse a ésta, disminuye la síntesis de PTH
nto, inhibe la síntesis y secreción de la PTH
El receptor del Factor de Crecimiento Fibroblástico (R-FCF) y la proteína transmembrana “Klotho” forman un complejo que es estimulado por el Factor de Crecimiento Fibroblástico 23 (FCF-23). El FCF-23 es una hormona que se produce en los osteocitos y osteoblastos cuando aumentan los niveles de fosfato y de calcitriol. El FCF-23 inhibe la secreción de PTH en las glándulas paratiroideas y disminuye la reabsorción de fosfato en el túbulo proximal del riñón.
La PTH actúa en el hueso sobre los osteocitos, osteoclastos y osteoblastos, incrementando los niveles de calcio. En el riñón promueve la retención de calcio y excreción de fosfato, y en el intestino, estimula la absorción del calcio.
A nivel del túbulo proximal renal, normalmente se realiza un intercambio recíproco entre el calcio y el fósforo para mantener sus niveles estables. Así mismo la 1α-hidroxilasa permite la activación de la vitamina D (calcitriol). La acción de la 1α-hidroxilasa es suprimida por el FCF-23. La hipocalcemia e hiperfosfatemia estimulan la producción de PTH, mientras la vitamina D la inhibe 6,7,8(tabla 1).
Fisiopatología y causas del hiperparatiroidismo primario
En el HPTP el trastorno metabólico se localiza en las glándulas paratiroideas por pérdida en el control de la síntesis y secreción de la PTH. En más del 80 % de los casos se detecta hipercalcemia asintomática, asociada a elevación de la PTH. Sin embargo, la hipercalcemia también puede presentarse con valores normales o mínimamente elevados de PTH. El HPTP normocalcémico 8 es una de las formas de presentación del HPTP, en el que existen, en pruebas repetidas, niveles elevados de PTH con calcio sérico normal. Para su diagnóstico, se deben excluir otras causas de hiperparatiroidismo secundario, como enfermedad renal, deficiencia de vitamina D, medicamentos o síndromes de malabsorción, entre otros.
La hipercalcemia hipocalciúrica familiar (HHF), una condición benigna hereditaria, es uno de los diagnósticos diferenciales principales del HPTP. Se presenta en individuos jóvenes, con hipercalcemia crónica y niveles de PTH normales o levemente elevados 9. Según las guías clínicas, el cociente de aclaramiento calcio/creatinina (CCCR) es el índice bioquímico de elección para diferenciar entre HPTP y HHF. Un cociente menor de 0,01 es sugestivo de HHF y uno mayor de 0,02, de HPTP. Los pacientes con HHF no requieren tratamiento quirúrgico 10.
En aproximadamente el 86 % de los casos de HPTP, la causa es un adenoma único de paratiroides, en el 9 % hiperplasia difusa o nodular de una o varias glándulas, en el 3 % adenoma doble y en el 1-2 % carcinoma.
Cuando se trata de un carcinoma, generalmente hay órganos blanco comprometidos, por lo que se presenta osteoporosis, fractura patológica, urolitiasis, pancreatitis, o trastorno de la conciencia. En el 75 % de los casos el nivel de calcio sérico está por encima de 14 mg/dl y la PTH intacta elevada más de 4 veces su valor normal. Puede cursar con parálisis del pliegue vocal, en el 50 % de los casos se palpa nódulo en el cuello y en el 75 % su tamaño es mayor de 2 cm 11,12.
Otras causas de Hiperparatiroidismo primario
Síndromes hereditarios
En un 95 % de los casos, el HPTP es de presentación esporádica 13; sin embargo, en muy raras ocasiones se presenta como parte de síndromes familiares, en edades tempanas. El hiperparatiroidismo familiar incluye un grupo de desórdenes en donde el HPTP es heredado, usualmente, como una característica autosómica dominante.
El síndrome de neoplasia endocrina múltiple (NEM) tipo 1, es causado por mutaciones inactivadoras en el gen NEM1 (11q13) que codifica la proteína menina, un supresor tumoral. Inicialmente descrito en 1954, es la causa más común de HPTP familiar, representando aproximadamente 2-4 % de todos los casos 13. Se caracteriza por una predisposición a desarrollar tumores endocrinos a nivel de la hipófisis, paratiroides y tumores neuroendocrinos pancreáticos y del tracto gastrointestinal. La asociación de la mutación del gen NEM1 con la presentación esporádica o familiar de adenomas paratiroideos ha sido bien documentada, mientras que su asociación con el carcinoma paratiroideo es rara. El HPTP es el trastorno endocrino más frecuente, presente en alrededor del 90 % de los pacientes entre 20 y 25 años y generalmente hay compromiso multiglandular con crecimiento de todas las glándulas.
La NEM tipo 4, más recientemente descrita, es similar al NEM1. Los afectados desarrollan tumores paratiroideos, hipofisiarios, pancreáticos, adrenales y, en raras ocasiones, de cérvix y testículos. Es causado por mutaciones inactivadoras en el gen CDKN1B, que codifica la proteína p27, la cual actúa como un regulador de la progresión del ciclo celular. Más de un 80 % de los afectados por este síndrome, presentan hiperparatiroidismo primario 14.
El síndrome NEM tipo 2A, causado por mutaciones en el protooncogen RET en el cromosoma 10, se caracteriza por el hallazgo de carcinoma medular de tiroides, feocromocitoma e hiperplasia o adenoma de la paratiroides. El HPTP ocurre en un 20-30 % de los casos, usualmente con presentación clínica leve o asintomática. La progresión maligna de los adenomas paratiroideos es rara 13.
El síndrome de hiperparatiroidismo por tumor mandibular (HPT-TM) es causado por mutaciones en el gen HPRT2 en el cromosoma 1, que originan tumores paratiroideos, fibromas osificantes de la mandíbula, lesiones renales (tumor de Wilms, carcinoma papilar renal, enfermedad quística) e HPTP 14. En más del 95 % de los pacientes, la primera manifestación es el hiperparatiroidismo primario, con un comportamiento más agresivo por hipercalcemia severa y con frecuencia inusualmente alta de carcinoma paratiroideo (10-15 %) 13.
Otro de los síndromes hereditarios, es el Hiperparatiroidismo Familiar Aislado, muy poco frecuente, que se caracteriza por la presencia de hiperparatiroidismo primario causado por tumores únicos o múltiples paratiroideos, en al menos 2 familiares de primer grado y en ausencia de otros tumores o trastornos endocrinos. No se conoce el mecanismo especifico, pero se han detectado mutaciones en el gen NEM1 (20-23 %), en el gen RECa (receptor especifico de calcio, en 14-18 %) y en el gen HRTPT2, en menor porcentaje (tabla 2).
Tabla 2. Características de síndromes hereditarios en hiperparatiroidismo primario.
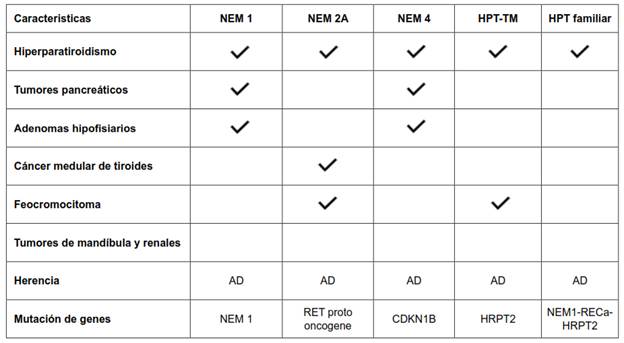
NEM:* neoplasia endocrina múltiple, HPT: hiperparatiroidismo, HPT-TM: hiperparatiroidismo - tumor mandibular, AD: autosómico dominante, CDKN1B: inhibidor 1B de quinasa dependiente de ciclina, RECa: receptor específico del calcio.
Exposición a radiación
Pacientes con HPTP pueden tener historia de irradiación a cabeza y cuello, en un rango de 20 a 40 años previos al desarrollo de la condición. El ejemplo más representativo, es una cohorte de 61 trabajadores de la planta nuclear en Chernóbil en 1986 donde 15 de ellos desarrollaron HPTP. La exposición media fue entre 0,3 a 8,7 Gy. No se ha demostrado diferencias en el curso clínico o recurrencia entre pacientes expuestos y no expuestos. Sin embargo, en pacientes expuestos es más frecuente la asociación con tumores tiroideos 15.
Medicamentos
La terapia con litio y diuréticos tiazídicos se han asociado como factores de riesgo para el HPTP 16. La experiencia más reciente con las tiazidas ha sugerido que la hipercalcemia, en este contexto, posiblemente enmascara el estado subyacente del HPTP, y no es probable que se revierta cuando se suspende el diurético 17.
Diagnóstico
La mayoría de los casos de HPTP son diagnosticados incidentalmente por laboratorios de rutina o dentro del seguimiento de osteopenia o urolitiasis. Clínicamente, se presenta como hipercalcemia asintomática en alrededor de 80 % de los casos.
El HPTP sintomático puede manifestarse con dolores óseos, fatiga, pérdida de peso, úlcera péptica, pancreatitis, nefrolitiasis, arterioesclerosis acelerada, déficit cognitivo y ansiedad.
Los laboratorios frecuentemente muestran elevación del calcio y PTH séricos, y aumento en la excreción urinaria de calcio, y en las radiografías óseas se observa el patrón típico de “gránulos de sal y pimienta”, compatible con osteítis fibrosa quística, osteopenia del tercio distal de la clavícula, reabsorción subperióstica de las falanges distales, quistes óseos o tumores pardos.
La gammagrafía realizada con Tc99m(tecnecio)-Sestamibi, tanto en imágenes planares como de tomografía computarizada por emisión de fotón único (SPECT, por sus siglas en ingles), tiene utilidad en la localización de adenomas paratiroideos, especialmente en aquellos de tamaño superior a 500 mg 18,19. La sensibilidad y la especificidad mejora cuando las imágenes de SPECT son fusionadas con imágenes de la tomografía computarizada (figura 1), preferiblemente adquiridas en equipos híbridos, pasando la sensibilidad de 83 % a 96 % y la especificidad de 80 a 93 %, ventajas más evidentes en adenomas de menor tamaño, de alrededor de 210 mg 20. La sensibilidad es menor (58 %) en el caso de glándulas hiperplásicas 21. Cuando las imágenes de medicina nuclear convencional (SPECT) son negativas, una alternativa es realizar una tomografía por emisión de positrones (en inglés, PET-CT) con C11-Metionina, con F18-Colina o C11-Colina 22,23.
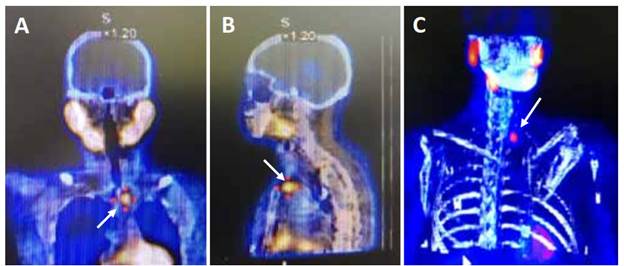
Figura 1. Gammagrafía de paratiroides con SPECT-CT que muestra hipercaptación de la glándula paratiroidea inferior izquierda (flechas blancas).
Es recomendable tener una ecografía de tiroides para descartar patología tiroidea concomitante. Otras imágenes como la tomografía contrastada, resonancia magnética, angiografía con muestreo selectivo de PTH o PETscan, sólo se utilizan en caso de gammagrafía negativa, recidiva o difícil localización de la glándula o las glándulas enfermas y de tejido paratiroideo ectópico 24.
Como se explicó previamente, un diagnóstico diferencial es la hipercalcemia hipocalciúrica familiar benigna, que cursa con niveles normales o ligeramente elevados de PTH. Generalmente se diagnostica por persistencia de hipercalcemia posterior a paratiroidectomía parcial 25.
Aunque se sospeche carcinoma primario de paratiroides, no es recomendable realizar biopsia con aguja fina, por su baja especificidad y por la posibilidad de desarrollar paratiromatosis.
Tratamiento
La cirugía es el tratamiento recomendado en el HPTP (tabla 3), para el paciente, sintomático o asintomático, que tenga compromiso de órgano blanco (osteopenia, urolitiasis, disminución de la tasa de filtración glomerular, úlcera péptica, pancreatitis o trastorno neuromuscular).

Un cirujano experimentado está en capacidad de identificar la o las glándulas afectadas en el 95 % de los casos. La fusión de imágenes (SPECT-CT) y la medición de la PTHi (molécula intacta) en el transcurso de la cirugía, han permitido tomar decisiones terapéuticas más acertadas, disminuyendo la tasa de recidiva del HPTP a menos del 5 % 26. La cirugía radioguiada y la decisión de realizar abordaje invasivo mínimo dependerá de la tecnología disponible en cada centro médico y de la experiencia del cirujano (figuras 2 y 3).
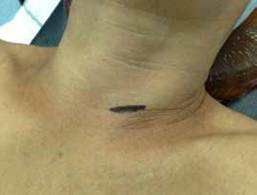
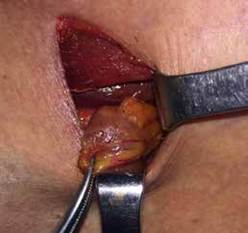
Figura 3. Adenoma de paratiroides inferior izquierda. a. Músculo esternotiroideo; b. lóbulo tiroideo izquierdo; c. adenoma de paratiroides.
Habitualmente es un procedimiento ambulatorio, el paciente egresa con suplementos de calcio (3 gr/día) y calcitriol (0.25 mcg/día). Se realiza control en 3 semanas con nuevos valores de PTHi, fosfatasa alcalina, calcio y fósforo séricos.
En pacientes de alto riesgo quirúrgico, o que rechazan el procedimiento, se debe recomendar un seguimiento anual estricto con niveles de calcio, creatinina y densitometría ósea. En algunos casos con hipercalcemia sintomática se recomienda mantener buena hidratación, restricción de la ingesta de calcio, asegurar niveles normales de vitamina D, e incluso, el uso de calciomiméticos. Es recomendable que la decisión terapéutica sea tomada entre el cirujano, el endocrinólogo y el paciente 27,28,29.
En el carcinoma primario se recomienda cirugía radical (previa corrección de la hipercalcemia severa), con hemitiroidectomía ipsilateral, exploración contralateral y hasta el timectomía, con linfadenectomía en caso de compromiso histológico documentado.
Complicaciones del manejo quirúrgico
La complicación más frecuente es la hipocalcemia transitoria, que se presenta entre el 15 % y el 30 % de los pacientes, y representa el 7 % de reingresos en postoperatorio para administración endovenosa de calcio. Cuando el paciente ingresa por urgencias y se documenta hipocalcemia sintomática, la administración de calcio debe ser simultáneamente oral y endovenosa. Se recomienda infusión de gluconato de calcio 2 ampollas en 100 cc de solución salina normal o en dextrosa en agua, para pasar en 15 minutos (no se debe usar lactato de ringer por la precipitación del gluconato), y continuar con 1 ampolla cada 6 horas hasta controlar los síntomas. El calcio oral se administra 1.5-3 gr cada 6 horas, en presentación de carbonato o citrato, y el calcitriol 0.25 mcg cada 8 horas. Con este manejo se logra el egreso del paciente en 12 a 24 horas en la gran mayoría de casos.
En algunos casos, la hipocalcemia postoperatoria es severa y prolongada, a pesar de tener una PTH normal o ligeramente elevada, y se puede asociar a hipofosfatemia, hipomagnesemia e hipercalemia, requiriendo manejo hospitalario con monitoreo cardiaco, suplemento oral y endovenoso de calcio, magnesio y calcitriol. Esta condición se conoce como “síndrome del hueso hambriento” y se presenta en aproximadamente un 13 % de los pacientes llevados a paratiroidectomia por HPTP. Generalmente afecta a pacientes con osteítis fibrosa y a pacientes que han recibido terapia con calciomiméticos por un tiempo prolongado. También se consideran factores de riesgo para desarrollarlo, la edad menor de 45 años, obesidad, niveles altos de fosfatasa alcalina y niveles normales o bajos de calcio sérico preoperatorios 8,30,31. En instituciones con alto volumen de cirugía de paratiroides y con recursos, han utilizado la criopreservación de tejido paratiroideo con el fin de reimplantarlo en caso de hipoparatiroidismo severo de difícil manejo 32.
El sangrado, la infección o la lesión del nervio laríngeo recurrente se presentan aproximadamente en el 1 % de los casos. La mortalidad perioperatoria es del 0,3 % 33,34,35.
La complicación menos deseada por el cirujano es la imposibilidad de encontrar la o las glándulas enfermas y la consecuente persistencia del hiperparatiroidismo. Generalmente las fallas se presentan en la localización preoperatoria, en la exploración cervical o por subestimar el número de glándulas paratiroideas con hiperplasia versus adenomas. En la serie reportada por Tominaga Y., informó una incidencia de HPT persistente del 4% y una tasa de reintervención por esta causa, del 1,6 % 35.
La paratiromatosis es la siembra de múltiples focos de tejido paratiroideo benigno e hiperfuncionante en los tejidos blandos del cuello o mediastino, producto de la ruptura de la cápsula de la glándula paratiroidea durante la exploración quirúrgica, de la realización de biopsia con aguja fina, o del uso percutáneo de la escleroterapia con alcohol. Estos dos últimos procedimientos no son recomendados de rutina 35.
Conclusiones
El hiperparatiroidismo primario, es una causa común de hipercalcemia. Su presentación clínica ha evolucionado hasta una condición mayormente asintomática en la población, cuyo diagnóstico se hace a partir de exámenes bioquímicos rutinarios. En pacientes jóvenes, su abordaje diagnóstico debe ser más cuidadoso; en estos casos puede ser un componente de múltiples anormalidades endocrinológicas hereditarias que, si bien representan menos del 5 % del HPTP, afectan con mayor frecuencia a este grupo etáreo. La cirugía es el único tratamiento curativo de esta enfermedad; se recomienda en pacientes jóvenes sintomáticos, o en aquellos asintomáticos que están en riesgo de presentar compromiso de órgano blanco. En la actualidad, el estudio prequirúrgico con imágenes diagnósticas (gammagrafía o SPECT) permite localizar el adenoma con mayor precisión, evitando recidivas y permitiendo abordajes de invasión mínima.

