










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Cuidarte
Print version ISSN 2216-0973
Rev Cuid vol.8 no.1 Bucaramanga Jan./Apr. 2017
https://doi.org/10.15649/cuidarte.v8i1.373
Articles
APLICACIÓN DE LA ALEATORIZACIÓN MENDELIANA: ¿PODEMOS ESTABLECER FACTORES DE RIESGO CAUSALES PARA LA DIABETES TIPO 2 EN PAÍSES DE BAJO Y MEDIO INGRESO?
1BSc, Integrative Epidemiology Unit, University of Bristol, Bristol, United Kingdom. Corresponding author, E-mail: ryan.langdon@bristol.ac.uk
2 PhD, Integrative Epidemiology Unit, University of Bristol, Bristol, United Kingdom, E-mail: kaitlin.wade@bristol.ac.uk
La carga mundial de diabetes tipo 2 (DT2) está en aumento, parcialmente facilitada por un marcado incremento de la enfermedad en países de bajos y medianos ingresos (PBMI)1,2. Los PBMI no solo muestran una alta prevalencia de DT2 (8.7 %), pero han evidenciado un aumento más acelerado en esta prevalencia en los últimos 30 años comparados con los países de altos ingresos (PAI)1. Los factores de riesgo convencionales para DT2 en los PAI, como alto índice de masa corporal (IMC)3, bajos niveles de actividad física4 y malos comportamientos alimentarios5), no dan cuenta por completo del mayor aumento en la prevalencia de DT2 en los PBMI2. Por lo tanto, los factores de riesgo para DT2, específicamente dentro de un contexto de PBMI, deben ser determinados.
En la actualidad, se están utilizando análisis epidemiológicos observacionales para explicar la creciente prevalencia y determinar los factores de riesgo sobresalientes para DT2 en los PBMI, pero aun si están bien diseñados, dichos estudios son propensos a la confusión, causalidad inversa y múltiples fuentes de sesgo (por ejemplo, selección y medición)6. Como tal, estos diseños de estudio pueden potencialmente generar estimaciones poco fiables de causalidad entre un factor de riesgo y la enfermedad. Un enfoque exitoso y más robusto para superar estas limitaciones y mejorar la inferencia causal es la aleatorización Mendeliana (AM)6,7.
Brevemente, como resultado de los análisis de variable instrumental (VI) en econometría, La AM explota la primera y segunda leyes de herencia de Mendel (es decir, la distribución independiente y la segregación de alelos que conlleva a la distribución aleatoria de los genotipos en la población), permitiendo el uso de variantes genéticas para sustituir un rasgo clínicamente relevante (y usualmente modificable) (por ejemplo, IMC)6,7. Siempre y cuando se cumplan una serie de supuestos (Figura 1), estas VI genéticas pueden luego ser utilizadas para derivar el efecto causal sobre un rasgo en la enfermedad o resultado adverso para la salud, confiriendo múltiples ventajas sobre estimaciones de asociación derivadas de observaciones.
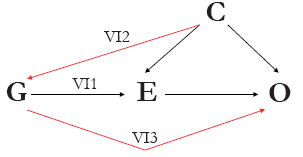
Figura 1 Gráfico acíclico dirigido de la teoría y supuestos clave de la aleatorización Mendeliana. Una variante genética (o variantes, G) se pueden utilizar como variables instrumentales para una exposición de interés (E) para evaluar la asociación causal entre E y el resultado de interés (O), dado que los siguientes tres supuestos se mantienen: (VI1) G debe ser ampliamente asociada con E; (VI2) G no debe ser asociada con ninguna variable de confusión medida o no medida (C); y (VI3) no debe haber asociación independiente entre G y O, dado E y C.
Análogas a los brazos de un ensayo clínico aleatorizado (ECA), las variantes genéticas utilizadas en la AM son ampliamente independientes de los factores de confusión debido a la naturaleza aleatoria de su asignación dentro de una población. Éstas tampoco son modificadas por el consiguiente desarrollo de la enfermedad o resultado de la salud (Figura 2) y, con el advenimiento de matrices de genotipificación más precisas, se reduce sustancialmente el error de medición. Por lo tanto, a nivel poblacional, la porción de varianza en el rasgo modificable explicado por las variantes genéticas utilizadas como una VI (contrario a la medición directa del rasgo) está libre de las limitaciones que de otro modo debilitaría la inferencia causal en estudios observacionales. La AM proporciona una estimación robusta y sin confusión de la asociación causal entre un rasgo y la enfermedad6,7.
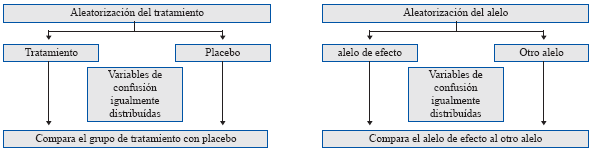
Figura 2 Metodología de aleatorización Mendeliana comparada a ensayos clínicos aleatorios (ECAs). En un ECA, los individuos se asignan al azar a los brazos de tratamiento o de control (placebo), lo cual teóricamente asegura que los factores de confusión sean igualmente distribuidos entre los brazos del ensayo. Análogamente a esto, la asignación aleatoria y la distribución independiente de alelos en la concepción y la meiosis, respectivamente, asegura que los factores de confusión sean igualmente distribuidos entre los grupos de genotipos. Las diferencias en los individuos entre grupos de genotipos sólo se diferenciarán debido al grupo de genotipo; por consiguiente, podemos utilizar variantes genéticas como variables instrumentales para exposiciones de interés en los análisis de aleatorización Mendeliana.
Dentro del contexto de los PAI, la AM ha confirmado e identificado los factores de riesgo endógenos para DT2, incluyendo la obesidad, inflamación sistémica y lípidos en la sangre, y los factores de riesgo exógenos, incluyendo consumo de alcohol y lácteos8,9. Por ejemplo, el uso de la AM en una evaluación exhaustiva reciente de la asociación causal entre el IMC y la DT2 produjo estimaciones causales refinadas, que sugieren que el riesgo para DT2 aumentó aproximadamente 30% con cada unidad (kg/m2) en el IMC (odds ratio (OR): 1.26; intervalo de confianza (IC) del 95 %: 1.19; 1.32; P = 3,29 x 10-10)10. Sin embargo, estos hallazgos no pueden traducirse fiablemente a los PBMI. Hasta recientemente, el costo y la disponibilidad de grandes muestras de datos genéticos y fenotípicos restringieron el análisis de la AM a los PAI; una mayoría de los cuales residen dentro de poblaciones de ascendencia europea. Por tanto, aunque es probable que una mayor adiposidad puede, al menos en parte, explicar la creciente prevalencia de la diabetes en los PBMI, la estimación de la asociación no se ha probado y, actualmente, sólo existen unos pocos estudios de AM que evalúan el impacto de cualquier exposición modificable sobre riesgo de enfermedad especialmente en los PBMI.
Específicamente dentro del contexto de Latinoamérica, Borges et al.11 investigaron el efecto causal de la circulación de los niveles de homocisteína en la presión arterial en la Cohorte de Nacimientos de Pelotas de 1982 en Brasil utilizando la AM, luego compararon estos resultados a los de una cohorte de individuos europeos. Para el análisis europeo, los autores utilizaron datos resumidos de un meta-análisis reciente de estudios de asociación de todo el genoma (GWAS, por sus siglas en inglés)12, de homocisteína, (>44.000 individuos europeos) para generar una VI para los niveles circulantes de homocisteína, los cuales se utilizaron para comprobar el efecto causal de la homocisteína sobre la presión arterial (PA) utilizando datos resumidos del Consorcio Internacional sobre Presión Arterial (International Blood Pressure Consortium, IBCP) (>69.000 individuos europeos)13, en un enfoque de AM de dos muestras. Los resultados de AM demostraron que la presión sanguínea sistólica (PSS) disminuyó por 1.8 mmHg (IC 95 %: -3.9; 0.4; P = 0.11) en la cohorte de Pelotas, pero aumentó por 0.6 mmHg (IC 95 %: -0.8; 1.9; P = 0.41) en la población europea con cada desviación estándar (DE) aumenta en los niveles circulantes de log(homocisteína). Igualmente, la presión arterial diastólica (PAD) aumentó por 0.1 mmHg (IC 95 %: -1.5; 1.7; P = 0.93) en la cohorte de Pelotas, pero aumentó por 1,1 mmHg (IC 95 %: 0.2; 1.9; P = 0.01) en la población europea.
En otro análisis de AM utilizando la misma Cohorte de Nacimientos de Pelotas de 1982, Hartwig et al.14, encontró que la persistencia de la lactasa (es decir, consumo de leche) en adultos fue asociada de manera positiva con el IMC (efecto estimado por aumento de 1 dL/día en la ingesta de leche: 0.17 kg/m2; IC 95 %: 0.07; 0.27; P = 0.001) y riesgo de obesidad (OR por aumento de 1 dL/día en la ingesta de leche: 1.09; IC 95 %: 1.02; 1.17; P = 0.015), contrario a las estimaciones observacionales en la misma cohorte. La ingesta de leche en Brasil (y potencialmente otros países similares de Latinoamérica) puede predisponer a las personas a presentar un nivel más alto de adiposidad, un resultado que difiere de las estimaciones observacionales inconsistentes derivadas de las poblaciones europeas.
Estos dos ejemplos por sí solos subrayan la necesidad de que el análisis de AM se lleve a cabo en los PBMI, y los asuntos confrontados al tratar con datos genéticos. Primero, se requieren grandes tamaños de muestra para un poder estadístico adecuado en el análisis de AM debido a la, usualmente, relativamente pequeña porción de la varianza explicada en el factor de riesgo por la VI genética. El anterior ejemplo descrito, que evalúa el efecto causal de la circulación de niveles de homocisteína en la PA, destaca este punto particular. Además, un reciente GWAS en más de 340.000 personas encontró que 66 variantes genéticas asociadas con PA en los europeos también fueron predictivos de PA en 64.000 muestras no europeas; sin embargo, se necesitaron tamaños de muestra no europeos comparativamente grandes para mostrar esta concordancia entre la dirección de los efectos estimados y dilucidar cuáles variantes genéticas se asociaron con un grupo de ascendencia específico15. Por lo tanto, el tamaño de muestra notablemente diferente entre la Cohorte de Nacimientos de Pelotas de 1982 (N= 3.701) y el IBPC (N >69.000) en el estudio mencionado, puede explicar, en parte, la dirección opuesta de efecto generado entre las dos poblaciones. El costo relativo de las matrices de genotipado y procesamiento presenta un riesgo de poder estadístico limitado debido a tamaños de muestra más pequeños en el análisis de la AM, particularmente en los PBMI, donde las muestras de tamaño apropiado pueden ser relativamente escasas.
Segundo, los PBMI típicamente poseen una estructura poblacional multiétnica (como se vio en la Cohorte de Pelotas en los ejemplos anteriormente descritos), mostrando un alto nivel de mezcla genética y heterogeneidad. El efecto de los factores de riesgo en las enfermedades que típicamente afectarían una población ancestral (como europeos, por ejemplo) podría ser irrelevante para una población de un PBMI como resultado de esto. En un estudio reciente por Zanetti et al.16, las estimaciones de efectos globales para la asociación entre SNPs (polimorfismos de nucleótido único) y varias enfermedades comunes, como DT2, estaban típicamente en la misma dirección entre diferentes poblaciones ancestrales. Sin embargo, la estructura variable de desequilibrio de ligamiento (es decir, asociación no aleatoria de las variantes genéticas) entre diferentes ascendencias influye en gran medida la magnitud del efecto entre estas poblaciones. Por último, la diferencia en la magnitud de las estimaciones puntuales y probable heterogeneidad de la población en los países de Latinoamérica subraya el grado en que el análisis de la AM realizada en las poblaciones europeas puede no representar los efectos de las mismas exposiciones en los resultados en las poblaciones de los PBMI.
Aunque ningún estudio ha evaluado la relevancia causal de cualquier exposición en DT2 en los PBMI mediante el uso de la metodología de AM, los ejemplos de AM descritos no sólo resaltan su factibilidad en los PBMI, pero también sugieren que dichos estudios proporcionarán estimaciones de efecto más pertinentes a los PBMI. A medida que se publican más estudios sobre la AM en los PBMI, éstos podrían potencialmente refinar las VI específicas a la población que tienen en cuenta los antecedentes ancestrales, los efectos estimados y los factores de riesgo causales para enfermedades relevantes, como DT2.
Para que el análisis futuro de AM en los PBMI sea eficaz, hay ciertas propiedades, principios y limitaciones que se deben tener en cuenta, que ya han sido esbozadas y discutidas en detalle previamente6. Particular para los escenarios de los PBMI, la mezcla y la heterogeneidad de la población debido al origen multiétnico pueden introducir confusión genética y producir resultados sesgados. Esto se puede abordar utilizando métodos de control genómico ancestral implementados en GWAS. Además, el sesgo de selección y la generalización de los hallazgos (especialmente en poblaciones en las que es probable que exista un sobre-muestreo en grupos de bajo nivel socioeconómico) revisten especial importancia en la práctica. Finalmente, tales estudios también dependerán de la disponibilidad de datos genéticos, alto poder computacional e instalaciones de infraestructura apropiadas para almacenar, mantener y analizar los datos requeridos para el análisis de la AM.
Durante la última década, la metodología de AM ha sido ampliamente aplicada para mejorar la inferencia causal dentro de un rango de contextos epidemiológicos. Los recientes desarrollos metodológicos basados en el concepto básico de la AM, como AM de dos muestras, dos etapas, en red y AM multi-fenotipo han hecho tales análisis complejos fácilmente accesibles para la comunidad de investigación, y han proporcionado más herramientas para disecar redes causales entre rasgos con mayor poder estadístico (Tabla 1)6,7.
Tabla 1 Conceptos metodológicos, desarrollos recientes, fortalezas y limitaciones de los análisis de la aleatorización Mendeliana

Dada la alta prevalencia de DT2 en Latinoamérica y los PBMI, es importante generar mayor comprensión sobre los factores de riesgo potencialmente modificables para DT2 (junto con otras enfermedades de alta prevalencia y resultados adversos para la salud). Con el crecimiento económico, la disponibilidad de tejido humano, aumento de la costo-efectividad de las matrices de genotipificación y los recientes desarrollos en la metodología de la AM, los análisis de la AM se hacen cada vez más prácticos y pueden resultar de fundamental importancia al tratar de hallar factores de riesgo sobresalientes para DT2 que son particularmente pertinentes en los PBMI.
REFERENCES
1. NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet. 2016; 387 (10027): 1513-30. http://dx.doi.org/10.1016/S0140-6736(16)00618-8. [ Links ]
2. Dagenais GR, Gerstein HC, Zhang X, McQueen M, Lear S, Lopez-Jaramillo P, et al. Variations in Diabetes Prevalence in Low-, Middle-, and High-Income Countries: Results From the Prospective Urban and Rural Epidemiological Study. Diabetes Care. 2016; 39(5):780-7. http://dx.doi.org/10.2337/dc15-2338. [ Links ]
3. Prospective Studies Collaboration, Whitlock G, Lewington S, Sherliker P, Clarke R, Emberson J, Halsey J, et al. Body-mass index and cause-specific mortality in 900000 adults: collaborative analyses of 57 prospective studies. Lancet. 2009; 373(9669):1083-96. http://dx.doi.org/10.1016/S0140-6736(09)60318-4. [ Links ]
4. Zaccardi F, O'Donovan G, Webb DR, Yates T, Kurl S, Khunti K, et al. Cardiorespiratory fitness and risk of type 2 diabetes mellitus: A 23-year cohort study and a meta-analysis of prospective studies. Atherosclerosis. 2015; 243(1):131-7. http://doi.org/10.1016/j.atherosclerosis.2015.09.016. [ Links ]
5. Ezzati M, Riboli E. Behavioral and dietary risk factors for noncommunicable diseases. N Engl J Med. 2013; 369(10):954-64. https://doi.org/10.1056/NEJMra1203528. [ Links ]
6. Haycock PC, Burgess S, Wade KH, Bowden J, Relton C, Davey Smith G. Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. Am J Clin Nutr. 2016; 103(4): 965-78. https://doi.org/10.3945/ajcn.115.118216. [ Links ]
7. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014; 15; 23(R1): R89-98. https://doi.org/10.1093/hmg/ddu328. [ Links ]
8. Sandhu MS, Debenham SL, Barroso I, Loos RJ. Mendelian randomisation studies of type 2 diabetes: future prospects. Diabetologia. 2008; 51(2):211-3. https://doi.org/10.1007/s00125-007-0903-x. [ Links ]
9. Swerdlow DI. Mendelian Randomization and Type 2 Diabetes. Cardiovasc Drugs Ther. 2016; 30(1):51-7. https://doi.org/10.1007/s10557-016-6638-5. [ Links ]
10. Corbin LJ, Richmond RC, Wade KH, Burgess S, Bowden J, Davey Smith G, et al. BMI as a modifiable risk factor for type 2 diabetes: refining and understanding causal estimates using Mendelian randomisation. Diabetes. 2016; 65(10): 3002-7. https://doi.org/10.2337/db16-0418. [ Links ]
11. Borges MC, Hartwig FP, Oliveira IO, Horta BL. Is there a causal role for homocysteine concentration in blood pressure? A Mendelian randomization study. Am J Clin Nutr. 2016;103(1):39-49. https://doi.org/10.3945/ajcn.115.116038. [ Links ]
12. van Meurs JB, Pare G, Schwartz SM, Hazra A, Tanaka T, Vermeulen SH, et al. Common genetic loci influencing plasma homocysteine concentrations and their effect on risk of coronary artery disease. Am J Clin Nutr. 2013; 98(3):668-76. https://doi.org/10.3945/ajcn.112.044545. [ Links ]
13. The International Consortium for Blood Pressure Genome-Wide Association Studies. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011; 478(7367):103-9. https://doi.org/10.1038/nature10405. [ Links ]
14. Hartwig FP, Horta BL, Davey Smith G, de Mola CL, Victora CG. Association of lactase persistence genotype with milk consumption, obesity and blood pressure: a Mendelian randomization study in the 1982 Pelotas (Brazil) Birth Cohort, with a systematic review and meta-analysis. Int J Epidemiol. 2016; 45(5): 1573- 87. https://doi.org/10.1093/ije/dyw074. [ Links ]
15. Ehret GB, Ferreira T, Chasman DI, Jackson AU, Schmidt EM, Johnson T, et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat Genet. 2016;48(10):1171-84. https://doi.org/10.1038/ng.3667. [ Links ]
16. Zanetti D, Weale ME. True causal effect size heterogeneity is required to explain trans-ethnic differences in GWAS signals. bioRxiv. 2016. http://dx.doi.org/10.1101/085092. [ Links ]
Recibido: 18 de Noviembre de 2016; Aprobado: 16 de Diciembre de 2016
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
