











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Neurofibromatosis type 1 (NF1) has been indisputably linked to neurofibromatosis type 2 (NF2). Both are autosomal dominant, hereditary, neurocutaneous disorders that have high rates of de novo mutation and a high risk of tumor development. However, they are clinically and genetically distinct diseases and should be considered as different entities. On the one hand, NF1 is a common disease that affects chromosome 17, shows clinical evidence in the skin and peripheral nervous system, and causes bone dysplasia. On the other, NF2 involves chromosome 22 and is considered a rare disorder with a relative scarcity of cutaneous manifestations and high-grade malignancy; it is also very rare (Table 1). 1
Table 1 Oncogenic risk and genetic localization in neurofibromatosis.
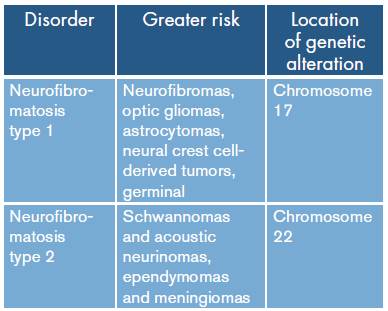
Source: Own elaboration based on 1.
NF1 is an autosomal dominant disorder that has a variable phenotypic expression, with manifestations ranging from moderate cutaneous lesions to severe orthopedic complications and functional alterations. 2 Its incidence is 1 per 3 000 live births and manifests clinically during childhood and adolescence. Half of the cases are sporadic, since no lesions are found in any of the parents; 90% of these mutations occur in paternal gametes. 3 The NF1 gene encodes a protein, neurofibromin, which acts as a tumor suppressor under normal conditions, regulating, in turn, another protein that stimulates cell growth and proliferation. In case of alteration, different tumor processes could be presented, such as the one observed in a small number of cases. 4,5
According to the World Health Organization, gliosarcoma is identified as a grade IV neoplasm. It was first reported in 1895 by Strobe, but it was not widely accepted until 1955 when Gross and Feigen described three patients with this type of lesion. 6 It is a mixed primary tumor of the central nervous system, composed of astrocytic anaplastic and malignant mesenchymal elements. 7 Furthermore, its represents between 1.8% and 8% of glioblastoma multiforme (GBM) cases. 8 It mostly affects supratentorial regions and is located in the temporal and parietal lobes, followed by the frontal and occipital lobes. 9-11 Regarding distribution by age and sex, it is observed more frequently in men, and its incidence increases significantly between ages 40 and 70; it is rare in adolescents and pediatric patients. 12,13 Its prognosis is similar to that of GBM, with a higher incidence of extraxial metastases being reported. 14 Regarding care, after establishing a possible diagnosis, surgery should be performed for characterization and as a therapeutic strategy for glioblastoma to relieve pressure and safely remove as much of the tumor as possible. Radiation therapy is almost always used, along with chemotherapy, after surgery or biopsy. The most commonly used chemotherapy drug in adults is temozolomide. Another area of interest for research has led to the development of techniques such as immunotherapy through vaccines or immunizations to treat this pathology. 15
CASE PRESENTATION
A 20-year-old male patient, of mixed race, from Popayán, Cauca, and with medium socioeconomic level attended the emergency service on March 7, 2016 due to intense headache and loss of postural tone. The patient reported history of NF1 only.
The patient showed a clinical picture of a month of evolution consisting of left hemicraneal headache, which did not yield to analgesics. He also presented impaired consciousness stupor, disorientation, preserved memory, dysarthria, loss of strength of the right half of the body, normal cranial nerves, decreased osteotendi-nous reflexes, and normal superficial and deep sensitivity. On admission, he presented two episodes of projectile emesis, while physical examination revealed pectum exacavatum, and multiple hyperpigmented skin lesions (café-au-lait spots) disseminated in the anterior region of the chest. Diagnostic studies were initiated and their academic importance was explained; the family understood and accepted said concept.
The patient was assessed by neurology, which requested a simple computed axial tomography (CT) of the brain, whose report suggested spontaneous intracerebral hemorrhage, described as a temporary lesion that displaces the midline and is accompanied by bleeding and perilesional edema (Figure 1). Various differential diagnoses were proposed, including brain tumor, glioblastoma, astrocytoma and middle cerebral artery aneurysm.
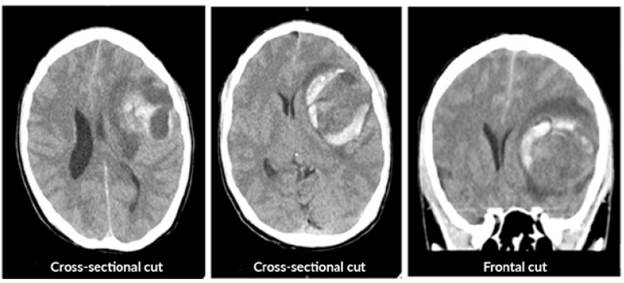
Source: Document obtained during the study.
Figure 1 Simple computerized axial tomography of the brain prior to surgery.
An emergency craniotomy for drainage was scheduled based on the results of the CT scan. Craniotomy showed a yellowish, hard tumor mass with necrosis and suggestive of astrocytoma. The patient did not require antibiotic management after the procedure and a new CT scan was requested, which showed satisfactory tumor resection with residual perilesional edema, without deviation from the midline (Figure 2). The histopathological study described resection compatible with glioblastoma (Figure 3).
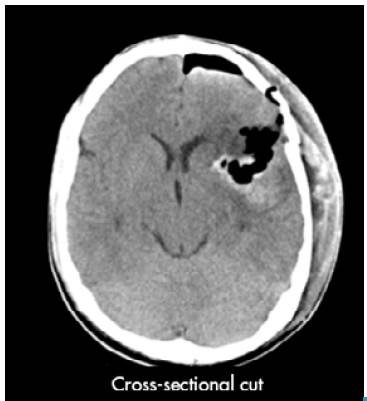
Source: Document obtained during the study.
Figure 2 Simple computed tomography of the brain after surgery.
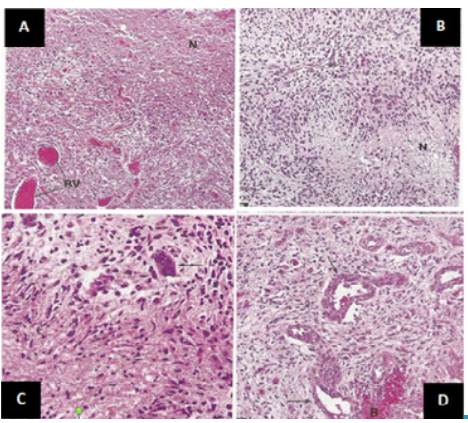
Source: Document obtained during the study.
Figure 3 Histopathological findings. A) high cellularity, abundant blood vessels and wide necrotic areas; B) prominent cellularity around a vessel and pseudoempalized cellularity around a necrotic area; C) variation in size and shape of the nuclei, some are markedly enlarged; D) small and medium blood vessels with thick walls that tend to bleed and endothelial cell proliferation.
Immunohistochemistry studies were performed to confirm diagnosis, describing diffuse positive mesenchymal component for vimentin CD99, very occasional for desmin and negative for smooth muscle actin. It was also necessary to find the positive component for GFAP, S100. Immunohistochemical staining for GFAP complemented reticulin staining by confirming the presence of two cell populations in gliosarcoma; it revealed epithelial spindle cell proliferations and intramural fusiform cells within thick-walled vessels stained for GFAP, S-100 protein and/or vimentin. The lesion was negative for estrogen receptors and occasional positive cells for progesterone receptors and EMA (epithelial membrane antigen) were found. The proliferation index measured by Ki67 was 60%, which confirmed the diagnosis of grade IV gliosarcoma.
During his recovery, the patient was he-modynamically stable, with motor, disharmonic and facial asymmetry that evolved satisfactorily with the help of phonoaudiological therapy. The patient was capable of returning to his daily activities after two weeks without any problem and continued receiving treatment with radiotherapy and chemotherapy.
DISCUSSION
Malignant gliomas represent 35-45% of all adult brain tumors and about 85% of them are glioblastomas; therefore, glioblastomas represent 29.7-38.2% of all brain tumors in adults. 12,16 In turn, gliosarcoma represents about 2% of all glioblastomas and 0.59-0.76% of all adult brain tumors 10; it is usually found in the supratentorial region with a slight preference for the temporal lobes 8, although it can affect the frontal, parietal or occipital lobes and corpus callosum less frequently. 17 Its clinical profile may include an intracranial hypertension syndrome such as headache, dizziness, vomiting, papilledema, seizures and motor deficit. CT usually shows a well-defined hyperdense lesion with marked perilesional edema, necrotic areas, and mass effect. 18
Feigin and Gross 19 were the first to demonstrate that gliosarcoma originates from the neoplastic transformation of blood vessels in a pre-existing glioblastoma. Currently, immunohistochemistry and genetic studies support that theory, suggesting a monoclonal origin for histological elements. 20
Immunohistochemical findings allow identifying the glial component of the glial fibrillary acidic protein (GFAP) and the S-100 protein. 14 Epithelial components include cytokeratins and immunoreactivity for p53 and, sometimes, actin if there is a muscular component. 21 GFAP immunostaining is more important to differentiate between gliosarcoma and glioblastoma, since it is found in the glial regions, although in very low amounts in sarcomatous regions. Vimentin is a marker of mesenchymal cells, with strong staining in sarcomatous areas. 22,23
Several treatment options are used to combat this form of brain cancer and the selected procedure depends on the location and severity of the tumor. In general, the treatment includes surgery, radiotherapy and chemotherapy with nitrosoureas, misonidazole, dacarbazine, mithramycin, ametophtherin, thalidomide, temozolomide, irinotecan, vincristine, cisplatin or doxorubicin. 16 The tumor can be removed surgically if its location is favorable for performing an extraction surgery and, usually, this procedure is followed by chemotherapy. Some research suggests that drugs such as temozolomide and bevacizumab (avastin) can be used to treat this pathology. 24,25
Some of the studies found show evidence of the interaction between patients with NF1 and gliosarcoma. Pathological characteristics (increased expression of p53) suggest that there is no overexpression of EGFR, as in primary glioblastomas, and that the increase of proliferation indexes could anticipate a bad prognosis in general. However, statistics report that the median overall survival of glioblastoma patients is 75% after 6 months and 19% after one year, and it is also better in patients with NF1. 4,26
This study had some limitations since the samples for immunohistochemistry tests were processed in a different laboratory and the delivery of the results took some time.
CONCLUSIONS
The early diagnosis of glioblastoma favors its timely management as connecting it with neurofibromatosis favors the prognosis of these patients. Joint management with surgery and radio and chemotherapy is required, since this favors the survival of the treated patients. Maintaining constant follow-up of patients helps detecting recurrences and facilitates subsequent resection and joint management. These patients need comprehensive and interdisciplinary management to achieve full rehabilitation.

