











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Amyloidosis comprises a heterogeneous group of diseases characterized by protein deposition (27 different types) that adopt a crossed β structure to form amyloid fibrils in the extracellular space. Primary systemic amyloidosis (amyloid light-chain, or AL) is one of them and is part of the spectrum of plasma cell neoplasms, in which an aberrant clone (<20% of plasma cells in the bone marrow) exaggeratedly produces immunoglobulin light chains (more frequently lambda), forming amyloid. 1 In the USA, the estimated annual incidence is 3 000 cases 2, the age at onset varies between the fourth and seventh decade of life, and is predominant in men. 3 Some patients may develop localized amyloidosis in the genitourinary or respiratory tract, in the lymph nodes or in the conjunctiva. 4,5
Its pathophysiology is still not completely clear: the damage is the result of mechanical interference and amyloid accumulation in the extracellular matrix of the vessels with apoptosis and ischemic damage. 6,3 The first tissues to be affected are blood vessels, which generates early endothelial microcirculatory dysfunction. 6 AL amyloidosis is associated in 10-15% of multiple myeloma cases and may be preceded by it or developed concomitantly 7; a similar proportion of patients with multiple myeloma will develop asymptomatic amyloid deposition. 3,8 In general, this pathology can affect any organ, except the brain 4, and the most common presentations are nephrotic syndrome, cardiomyopathy, peripheral sensory-motor neuropathy and hepatomegaly. 6,9 The following is the case of a patient with systemic AL amyloidosis and concomitant multiple myeloma who presented with respiratory failure and adynamic ileus.
CASE PRESENTATION
Male, mestizo patient, aged 47 years, from Bogotá D.C., meat vendor, who consulted for dyspnea and muscle weakness. Functional classification had decreased progressively for 18 months and worsened in the last 3 months, reaching the IV/IV classification on the NYHA scale, which is associated with orthopnea and lower limb edema. The patient presented with shoulder myalgia and gradual reduction of proximal muscle strength 11 months before consultation, with no paroxysmal nocturnal dyspnea. On examination, moderate to severe pulmonary hypertension and restriction in spirometry were found with mild oxygenation and hypercapnia disorder associated with obstructive sleep apnea-hypopnea syndrome (apnea/ hypopnea index: 8.7). Chronic thromboembolic pulmonary hypertension (CTEPH) was ruled out with ventilation/perfusion scan. Only class 1 obesity was reported as a pre-existing medical condition. The systems review revealed purplish eyelids and a mass in the perineal region -which increased with valsalva maneuvers-, abdominal distension, dysphagia to solids, constipation, erectile dysfunction and paresthesia of the hands.
On physical examination, the patient had a reading of 73% of arterial oxygen desaturation and positive purplish upper eyelids (raccoon eye) (Figure 1), class 2 jugular venous pressure, grade 2 systolic murmur in the tricuspid area, abdominal distension and decreased bowel sounds. A non-pruritic, non-painful tumor that bled easily was observed in the perineal skin (Figure 2). Grade II edema in lower limbs, decreased proximal muscle strength and single-breath counting of 1 2 (normal range >20) were also reported without muscle fatigability. Muscle hypertrophy of the bilateral supraspinus, bilateral deltoids and forearm muscles with pseudohypertrophy of paraspinal muscles were also evidenced.
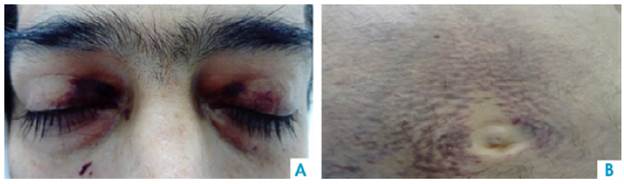
Source: Own elaboration based on the data obtained in the study
Figure 1 A. Periorbital ecchymosis. B. Perimumbilical purpuric macules.
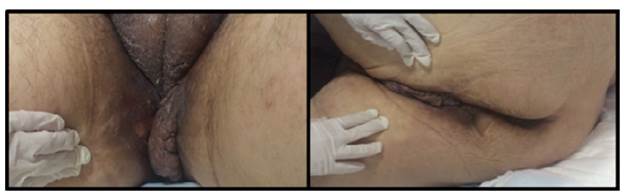
Source: Own elaboration based on the data obtained in the study.
Figure 2 Hyperpigmentation and perineal tumor, and scrotal infiltration.
On admission, myopathy studies were initiated (Table 1) and muscle enzymes, electromyography and muscle biopsy were requested, which excluded inflammatory myopathy. The autoimmune profile and HIV were negative. Adult Pompe disease was considered, but acid maltase was normal and syringomyelia was ruled out using contrast-enhanced cervical and brain MRI (Figure 3). Considering the lytic lesions observed in chest tomography, neoplasms (myopathy as a paraneoplastic phenomenon) were looked for as there was no compromise of the pulmonary parenchyma, only bibasal subsegmental atelectasis. No masses or organomegaly were observed in abdomen images (Figure 4).
Table 1 Summary of the main paraclinical tests of the case.
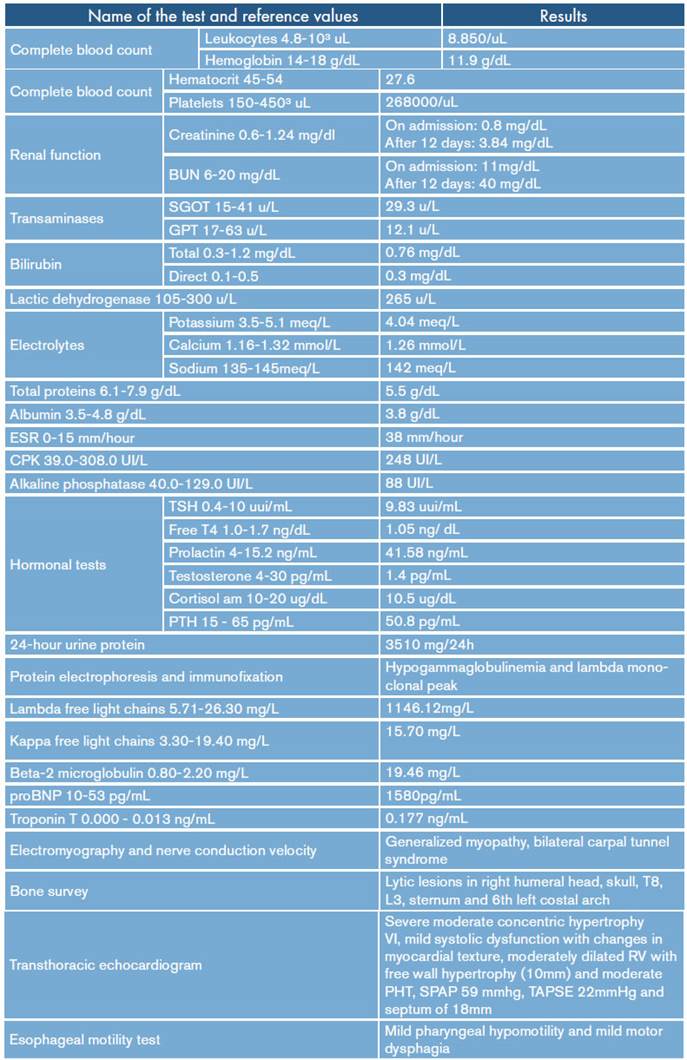
RV: right ventricle; LV: left ventricle; TAPSE: tricuspid annular plane systolic excursion; SPAP: systolic pulmonary arterial pressure; PH: pulmonary hypertension.
Source: Own elaboration.
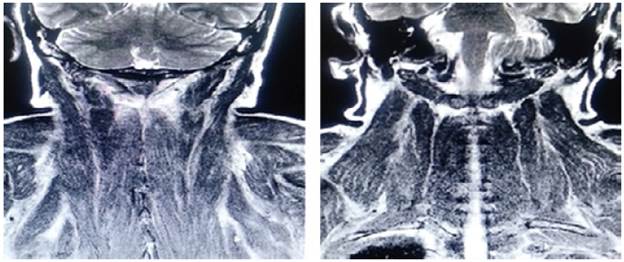
Source: Own elaboration based on the data obtained in the study.
Figure 3 Contrast-enhanced MRI of the head and neck, coronal plane, with paraspinal muscular edema.
Source: Own elaboration based on the data obtained in the study
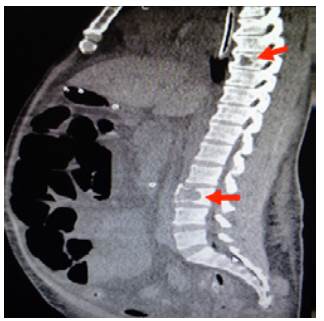
Figure 4 Sagittal CT scan of contralateral abdomen: distended loops and lytic lesions in T8 and L3 (arrows).
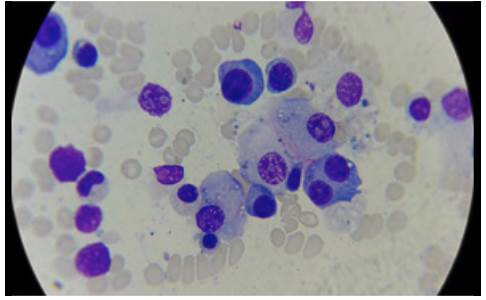
One week after admission, the patient presented acute kidney injury classified as stage 3 KDIGO (discarding prerenal origin, pharmacological toxicity and obstruction) and hypoxemic and hypercapnic respiratory failure, so he was transferred to the intensive care unit (ICU) for invasive mechanical ventilation and initiation of dialysis. Considering the presence of lytic bone lesions and hyperazoemia with nephrotic-range proteinuria by Bence Jones protein filtration, bone marrow aspiration and biopsy were performed, finding 80% of plasma cells (Figure 5). Lambda light chain multiple myeloma ISS III was diagnosed (Table 1). However, a colonoscopy was performed as the patient presented with myopathy, skin lesions, bilateral carpal tunnel, infiltrative cardiomyopathy, hypogonadism, erectile dysfunction and adynamic ileus that were not explained by the myeloma (Figure 6), finding extremely friable mucosa; perineal lesion and renal biopsy were taken to establish the presence of associated systemic amyloidosis.
Source: Own elaboration based on the data obtained in the study.
Figure 5 Bone marrow aspirate, 40x lens with plasmocytes, some atypical binucleated cells, eccentric nucleus, broad and basophilic cytoplasm.
Source: Own elaboration based on the data obtained in the study.
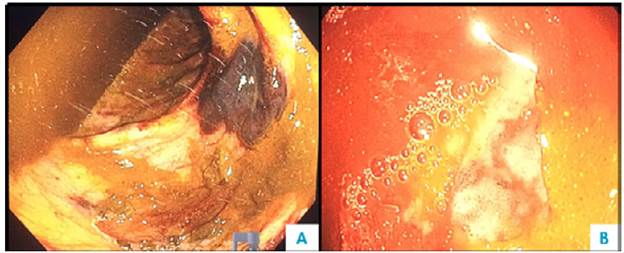
Figure 6 Colonoscopy. A) Submucosal hematoma and multiple mucosal ecchymoses; B) congestive mucosa and ulcer covered by fibrin.
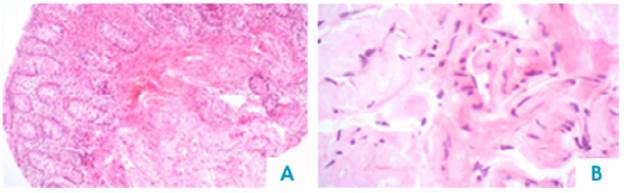
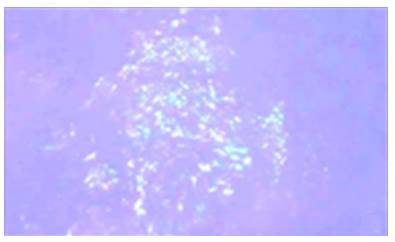
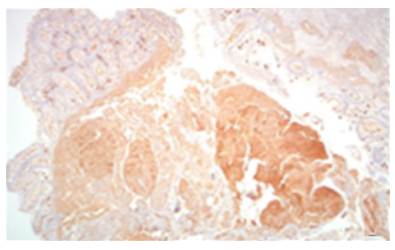
AL amyloidosis was confirmed by perianal lesion and colonic mucosa biopsy (Figures 7, 8, 9 and 10), where a deposit of eosinophilic amorphous material was found in the submucosa and vessel wall with the typical apple-green birefringence of Congo Red stained preparations under polarized light and confirmed with immunohistochemistry. Renal biopsy showed tubulopathy due to lambda free chain deposits, along with amyloid deposits in arterioles.
Source: Own elaboration based on the data obtained in the study.
Figure 7 Colon, submucosa and vessel wall biopsy with amorphous eosinophilic material deposit, using hematoxylin and eosin stain. A) 4x lens; B) 40x lens.
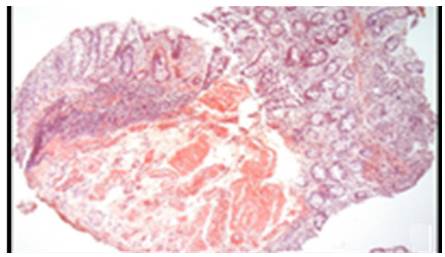
Source: Own elaboration based on the data obtained in the study.
Figure 8 Colon biopsy, Congo red staining with salmon-colored deposits in the thickened wall of the vessels. 4x lens.
Source: Own elaboration based on the data obtained in the study.
Figure 9 Colon biopsy, Congo Red staining. Apple-green birefringence on the wall of the glasses under polarized light with 40x lens.
Source: Own elaboration based on the data obtained in the study.
Figure 10 Colon biopsy, aspirated bone marrow. 40x lens with plasmocytes, some atypical binucleate cells, eccentric nucleus, broad and basophilic cytoplasm.
Chemotherapy following the CyBorD scheme was initiated due to the diagnosis of multiple myeloma with associated AL amyloidosis and the important systemic involvement in the patient. 20 days after the first cycle of chemotherapy, the subject was taken off dialysis. During the second cycle, bortezomib was discontinued and the dose of steroids was decreased to avoid potentiating its side effects (myopathy and neuropathy). Until that moment, the patient still depended on the ventilator due to a tracheostomy and required support pressure due to muscle fatigability. Adynamic ileus did not respond to multiple prokinetics nor neostigmine; enteral nutrition was maintained for trophic stimulation and total parenteral nutrition. After 4 months in the ICU connected to the ventilator with nutritional support and after four cycles of chemotherapy, the patient died due to ventilator-associated pneumonia and bacteremia due to Klebsiella pneumoniae, producer of carbapenemases.
DISCUSSION
The clinical case presented here is part of the 15% of AL amyloidosis cases associated with multiple myeloma. 7 Probably, amyloid deposits are present in patients with myeloma in a lesser proportion and such cases are not documented because there is no active search for these deposits. However, this case is relevant due to its clinical presentation, including severe involvement of multiple organs, onset with amyloidosis symptoms, development of myeloma with classic CRAB features on admission, and the diagnostic process represented by multiple differentials.
In a retrospective study conducted by the Mayo Clinic, of a total of 1596 patients, only 12 patients presented with myopathy, pseudohypertrophy, jaw claudication and creatine kinase concentration slightly increased to normal 6, so the specialists recommended measuring monoclonal protein during the evaluation of a patient with proximal non-inflammatory myopathy. 6 In a subsequent cohort of the same reference center, of 3 434 patients with AL amyloidosis treated between January 1995 and December 2015, 1.5% presented with muscle involvement, 22% myopathy only, 65% cardiac symptoms, 31% peripheral/ autonomic neuropathy, 25% renal symptoms, 8% liver symptoms and 4% gastrointestinal symptoms. 10
At the cardiac level, infiltration of the endocardium, atria and valves is found in amyloidosis, which generates contractile dysfunction due to restriction. 3,5 The most common early manifestation is dyspnea on exertion due to left ventricular diastolic dysfunction, which progresses to peripheral edemas and ascites. Atrial arrhythmia with thrombus formation 3, low blood pressure (due to decreased cardiac output and low peripheral tone), postural hypotension due to autonomic nerve disorder 3,5 and claudication of the jaw, legs and angina due to vascular involvement. For this, performing an electrocardiogram is recommended considering its low voltage, as well as an echocardiography with cardio-myocyte infiltration and magnetic resonance due to difficulty for draining the myocardium after gadolinium injection and a non-coronary pattern of increased gadolinium delay. Loop diuretics are the treatment of choice for this condition because angiotensin-converting enzyme inhibitors, angiotensin receptor blockers and beta-blockers are poorly tolerated due to hypotension. 3
Respiratory involvement, although rare, can occur with diaphragm or phrenic nerve infiltration; some cases have been reported. 11 In this case, significant muscle weakness with respiratory failure was observed, although a post-mortem diaphragm biopsy could not be performed. Pulmonary parenchymal, diffuse interstitial and tracheobronchial involvement, along with pulmonary hypertension type 1 by infiltration of the pulmonary vasculature, may also be observed. 11-13
The patient had gastrointestinal involvement (3.2% frequency) 2; nevertheless, colonoscopy showed ulcerations and friability of the mucosa, classic findings of amyloidosis together with thickening of the intestinal wall, polypoid protuberances, erosions and fine granular appearance of the mucosa. 14 Common symptoms are abdominal pain, esophageal reflux, constipation and nausea. Others include diarrhea, weight loss and early satiety, which may be caused by autonomic neuropathy, bacterial overgrowth or cardiac cachexia. Replacement of intestinal smooth muscle causes dysmotility, pseudo-obstruction and even ischemia secondary to vascular infiltration or ganglion cells depletion. 2 The most involved sites are the duodenum, the rectum and the esophagus. 5,15 Hepatomegaly may be a consequence of congestion or infiltration (by kappa chains, hard and non-pulsatile liver). 3,5 Management is symptomatic with antiemetics, prokinetics and nutritional support 15, but in this case, no medication worked.
Cutaneous and mucosal involvement is diverse; this patient presented with periorbital ecchymosis or "raccoon eyes", which is pathognomonic when associated with macroglossia. Other manifestations described were petechiae, ecchymotic macules, plaques, papules and nodules that simulate amber, hemorrhagic, normochromic or non-pruritic vesicles, which can appear in eyelids, retroauricular area, lips, tongue, oral mucosa, neck, armpits, submammary, navel and inguinal and anogenital area; the latter can simulate condylomata. 4 The perivascular amyloid deposit produces vessel fragility and spontaneous wounds or wounds caused by minimal trauma, as in this patient.
Although AL amyloidosis presents with nephrotic syndrome caused by hyaline deposits in the mesangium, in the glomerular basement membrane, in small arteries and in the tubular basement membrane, the reported case presented with "myeloma kidney" (tubulopathy caused by light chain deposits). Amyloidosis should be suspected in patients with myeloma and nephrotic proteinuria due to albuminuria, infiltrative cardiomyopathy, autonomic neuropathy, hepatomegaly and symptoms of partial intestinal obstruction. 16 Hypoadrenalism or hypothyroidism is less common 8, but this patient presented with subclinical hypo-thyroidism with hypogonadism. Differential diagnosis is wide, so the amyloid type should be confirmed, as well as other hematological malignancies such as lymphomas, waldenstrom macroglobulinemia and POEMS syndrome, which should be ruled out. 17
Diagnosis includes confirming paraproteinemia (around 90% of patients have it). Serum or urine electrophoresis sensitivity is approximately 50%, increasing to 80-90% with immunoelectrophoresis. 9 Histological confirmation by biopsy of the affected organ or subcutaneous fat has a sensitivity of 70-80%. 3 Hematoxylin and eosin show eosinophilic amorphous material, salmon red coloration in Congo red and apple green-birefringence in polarized light. Immunohistochemistry shows up to 92% amyloid subtype depending on the availability of antibodies with some limitation for the determination of AL amyloid, which has been attributed to difficulties, on the one hand, in the detection of conformational differences of light chains and the characteristics of the antibody used 18 and, on the other, the limited availability in some centers of a very sensitive method such as immunofluorescence for luminescent derivatives of polythiophene conjugates and immunoelectromicroscopy with gold-labeled antibodies and fibrillar antiproteins. 19 Currently, the gold standard is proteomic analysis of amyloid deposits by mass spectrometry, after microdissection of Congo red-positive deposits. 3
The prognosis of the disease depends on the number of affected organs, and there are prognostic biomarkers such as NT-proBNP and troponin T. The median survival is 6 months with cardiac involvement, but modern therapies based on bortezomib, dexamethasone and cyclophosphamide, followed by autologous stem cell transplantation, have shown longer survival rates with complete hematologic remission, unlike the first schemes with melphalan and prednisone 3,8, and are now the most commonly used schemes.
Likewise, there are reports of cases with localized involvement of the tracheobronchial vesicle that have been successfully managed by external beam radiotherapy. 20 The treatment of this systemic pathology is to suppress the plasmatic cell clone with chemotherapy and, in some cases, perform autologous bone marrow transplantation while supporting measures are taken to maintain the function of the organs involved as in the case of this patient. However, the progress of the disease, the prolonged stay in the ICU and the need for multiple devices favored his fatal outcome.
CONCLUSION
AL amyloidosis is a plasma cell dyscrasia that may be associated with multiple myeloma. Its onset spectrum is diverse, so a high index of suspicion should be considered so that it is not ignored, leading to an advanced stage that can be deadly. New chemotherapy schemes and timely diagnosis help improving survival.

