











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkINTRODUCTION
Neuromyelitis optica spectrum disorder (NMOSD) is a rare autoimmune disease in pediatrics with a chronic inflammatory demyelinating characteristic that affects the central nervous system (CNS) and may or may not be associated with seropositivity of anti-aquaporin-4 antibody (an-ti-AQP4 antibody). 1,2 4% of seropositive cases occur during childhood, at a female/male ratio of 3:1, and have a monophasic course. 2,3
The clinical presentations of NMOSD are optic neuritis and myelitis. The first causes a decrease in visual acuity from hours to days, pain with eye movement and blindness in one or both eyes, while the second may cause motor, sensory or autonomic involvement. 4-7
Early diagnosis and treatment, and relapse prevention are essential for improving the quality of life of patients with this disorder. The treatment of the acute form of this condition is mainly based on high doses of methylprednisolone and subsequent immunomodulatory management with azathioprine, mycophenolate or rituximab in some cases. 8-11 Most treatments are based on the management given to the adult population; therefore, clinical studies that address the pediatric population are required. 11
CASE PRESENTATION
15-year-old male patient, student, mestizo, from and resident of Bogotá D.C., belonging to a middle-income family, with a history of Hodgkin's lymphoma diagnosed at 4 years of age, treated with 5 rounds of chemotherapy and remission at age 5. No history of demyelinating neurological diseases in the family was reported.
The child attended a consultation on February 7, 2017 in the city of Tunja due to a slow and insidious clinical profile of 1 week of evolution consisting of hemiparesis on the right half of the body and loss of strength in upper limbs and trunk, which was associated with sensitive alteration at the cervical level and involvement of sphincters. Initially, the condition was investigated outside the institution as it was considered as a conversion syndrome; then, the patient was hospitalized and initial studies were performed to rule out a differential diagnosis of demyelinating disease. Contrast nuclear magnetic resonance (NMR) of the dorsal spine showed alteration of spinal cord signal intensity in the lower cervical segment, so complement studies were indicated.
That same day, the patient was admitted to the Clinica Universitaria Colombia in Bogotá D.C. due to suspicion of transverse myelitis based on a feeling of stabbing pain in the thoracic spine, associated with hemiparesis of the left side of the body with predominance in the upper limb, mild respiratory difficulty, hyporeflexia and non-voluntary loss of 2kg of weight in 1 week. A cervical spine MRI was performed, showing an alteration in spinal cord signal intensity in the cervical segment, correlated with the affected cervical sensory level. This finding allowed diagnosing longitudinally extensive transverse myelitis (LETM) (Figures 1 and 2), so the patient was admitted to the hospital to start therapy with methylprednisolone pulses at a dose of 1g intravenously every 24 hours for 5 days. In addition, immunophenotyping and diagnostic studies were performed, obtaining negative LETM.
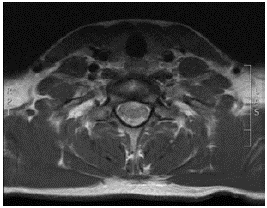
Source: Document obtained during the study.
Figure 1 Contrast magnetic resonance imaging (MRI) of the cervical spine (axial plane) that shows spinal cord injury involving anterior horns with abnormal contrast uptake and contrast uptake of the roots, mainly anterior C5, C6, C7 and C8.
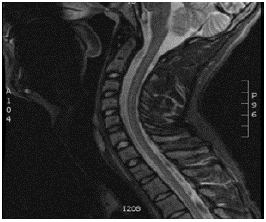
Source: Document obtained during the study.
Figure 2 Contrast nuclear magnetic resonance of the cervical spine (sagittal plane) that shows chronic changes in signal intensity and malacia in the anterior cords from level C2 to level C4.
The symptoms resolved with the therapy without associated adverse reactions, so he was discharged from hospital on February 27, 2017.
In April 2017, the patient received a new course of methylprednisone at a dose of 1g intravenously every 24 hours due to persistence of gadolinium enhancement in MRI of the cervical spine. On March 14, 2018, at 17:53 hours, he was readmitted to the Clinica Universitaria Colombia due to a clinical profile of 10 hours of evolution consisting of a decrease in bilateral visual acuity, associated with transient blurred vision, stabbing frontal and parietal headaches and a fall from his own height due to visual disturbance.
On physical examination during admission, the patient was alert, with bilateral 20/20 vision, normal bilateral fundoscopic exam, normal optical coherence tomography and without signs of focalization; the 4 limbs moved with 5/5 strength. Report of studies on cerebrospinal fluid (CSF) showed no pleocytosis or cellularity and normal lactic acid, ruling out encephalitis. The anti-AQP4 autoantibody in CSF taken on February 27, 2018 was positive: 4.53 U/mL with a cut point less than 3 U/mL (Figure 3). Additionally, it reported the presence of three standard oligoclonal bands type 2 in CSF. MRI of the brain and optical nerves following the demyelinating disease protocol were normal.
In the context of this patient, and given his pathological history, a relapse of this condition along with optic neuromyelitis was considered, reason why new boluses of systemic corticosteroid were indicated. Relapse of seropositive NMOSD was diagnosed because it met the criteria of anti-AQP4 autoantibody optical neuritis in positive CSF, acute myelitis with LETM, positive test for AQP4 and exclusion of other diagnoses. Therefore, he received a course of corticotherapy that started on March 15, 2018 for 5 days and was discharged from hospital with prescription of anti-CD20 therapy (Rituximab) at a dose of 375 mg/m2 of body surface in 4 weeks, with repetition every 6 months (dependent on titration of serum anti-CD20-CD27 every 9-10 weeks), to which he adhered properly.
The symptoms resolved and the disease stabilized thanks to the therapy prescribed, with adequate tolerance to the treatment. At the time of this case report, the boy feels that his condition is adequately controlled, with no associated severe adverse reactions, and has a favorable prognosis without further relapses since management with Rituximab was initiated.
This patient's timeline began with the onset of the symptoms in February 2017, a relapse with transverse myelitis in March 2017 and another relapse with optic neuromyelitis in March 2018, when the NMOSD diagnosis was confirmed. The findings of NMR of the brain ruled out other pathologies, mainly multiple sclerosis (MS) caused by transverse myelitis (not typical of MS) and absence of bright spot in T2 axial sequence. Anti-AQP4 autoantibody biomarkers in CSF were ordered based on these findings.
DISCUSSION
NMOSD, also known as Devic's disease, was first described in the nineteenth century as a monophasic disorder of severe acute transverse myelitis and bilateral optic neuritis. 1 This is a rare disease with a prevalence of 0.52-4.4 per 100 000 inhabitants and an incidence less than 0.05 cases per 100 000 inhabitants per year in the United States. 2,12 The average age of onset is 31 years, but some cases have been reported in the pediatric population. 3,4
The pathophysiological role of the disease is mediated by a blood cell autoantibody (NMO-IgG, or anti-AQP4 autoantibody) that binds to Aquaporin-4 (water channel found within the CNS) 5, specifically at the astrocyte junction with blood vessels. This antibody plays a key role in neuronal death by astrocytotoxic effect, mainly because of AQP4 dysfunction, complement activation and NK lymphocytes activation in regions with high density of AQP4 6-8 (Figure 1).
The clinical presentation of patients with NMOSD is highly variable and the symptomatology appears depending on the involvement of various areas of the CNS (optic nerve, spinal cord, area postrema, brain stem, diencephalon and brain). 9,10 The most frequent symptoms are longitudinally extensive optic neuritis, transverse myelitis and area postrema syndrome, consisting of vomiting or intractable hiccups. Other clinical manifestations that may occur with this disease derive from the involvement of the brainstem: vertigo, sensorineural hearing loss, facial paralysis, trigeminal neuralgia, diplopia, ptosis and nystagmus. 10,11,13 Symptomatic forms of narcolepsy or altered states of consciousness, encephalopathy associated with diffuse white matter injury in the CNS, posterior reversible encephalopathy syndrome and manifestations associated with dysfunction of the hypothalamic-pituitary axis such as SIADH have also been described. 14-15
Diagnostic criteria have9 changed over the years: an initial proposal was made in 1999 and then revised in 2006. The international consensus diagnostic criteria for NMOSD made a final proposal in 2015 16, as follows:
Diagnostic criteria for NMOSD with AQP4-IgG
1. At least 1 core clinical characteristic,
2. Positive test for AQP4-IgG using the best available detection method (cell-based assay strongly recommended),
3. Exclusion of alternative diagnoses.
Diagnostic criteria for NMOSD without AQP4-IgG or unknown AQP4-IgG status
1. At least 2 core clinical characteristics occurring as a result of one or more clinical attacks and meeting all of the following requirements:
At least 1 core clinical characteristic must be optic neuritis, acute myelitis with LETM, or area postrema syndrome,
Dissemination in space (two or more core clinical characteristics),
Fulfillment of additional MRI requirements, as applicable;
2. Negative test for AQP4-IgG using the best available detection method, or testing unavailable;
3. Exclusion of alternative diagnoses.
Core clinical characteristics
The same proposal included optic neuritis, acute myelitis, area postrema syndrome (episodes of hiccup or unexplained nausea and vomiting), acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic syndrome with typical NMOSD diencephalic lesions on MRI, and symptomatic brainstem syndrome with brain lesions typical of NMOSD on MRI as clinical characteristics of NMOSD.
Therefore, an imaging study that includes brain, orbits, single spine and contrast MRI must be performed for diagnosing NMOSD; anti-AQP4 antibody should also be determined. MRI findings are usually >2cm and located in periventricular zones of the third and fourth ventricles, that is, diencephalon and brainstem, supratentorial white matter, midbrain and cerebellum, which are areas where increased expression of AQP4 is observed. 8,9,17 Area postrema and hypothalamic lesions are usual in NMOSD with anti-AQP4 antibody, which is useful to differentiate it from other inflammatory and neurodemyelinating disorders of the central nervous system in the pediatric population. 9,18
The most common findings of spinal cord MRI is longitudinally extensive transverse myelitis, which is defined as a lesion that involves more than 3 contiguous vertebral segments and predominantly affects the central gray matter in the spinal cord. However, this finding is not as specific in children as it is in adults. 17-19
Differential diagnoses may include multiple infectious and reactive etiologies, among them, post-infectious and post-vaccinal etiologies, vascular pathologies, nutritional deficiencies - particularly, vitamin B12 deficiency-, compressive tumors, systemic lupus erythematosus, maculopathies and retinopathies. 4,8,11
Early diagnosis and treatment help prevent relapses, which are the origin of morbidity due to the neurodegeneration caused by this pathology. 4,12 Treatment should be provided early in order to reduce disability and morbidity 8,9,11,20; the therapeutic approach to the acute form of the condition is using high doses of methylprednisolone. 8,9,11 For relapse prevention, first-line immunomodulatory management includes azathioprine, mycophenolate, rituximab, among others; second-line includes metotrexate, mitoxantrone, and cyclophosphamide. 11,21-23
Regarding prognosis, more than half of the patients will continue to experience acute attacks that will result in possible and potentially permanent neurological disability. 23-24 About 80% of cases have a recurrent course of the disease and relapses are associated with long-term visual and motor disability. 11,19
This case is relevant due to the low incidence of the disease in pediatrics, the variability of the clinical presentation referred to in the literature, and onset with motor involvement that evolved to optical involvement and finally to seropositivity.
CONCLUSIONS
NMOSD is an entity with particular clinical, immunological and radiological findings that differentiate it from multiple sclerosis. Its diagnosis requires a high rate of suspicion, especially with presentations such as the one described with LETM. Early diagnosis (with a high rate of clinical suspicion) and early treatment help preventing relapses and major neurological sequelae (irreversible brain damage). More descriptive studies in pediatrics are needed to know the population and analytical studies to work on the treatment.

