











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Peroxisomes are organelles that use molecular oxygen to remove hydrogen atoms and produce hydrogen peroxide (H2O2) from organic substrates; these organelles are abundant in the cells of the nervous system, liver and kidney. Catalase is an enzyme found in the peroxisomal matrix that degrades hydrogen peroxide. Peroxisomes have multiple functions in humans such as the biosynthesis of plasmalogens, phospholipids and bile acids; a and (3 oxidation of fatty acids; pipecolic acid and phytanic acid oxidation; purine and polyamine catabolism; among others. 1
Peroxisomal diseases include peroxisomal biogenesis disorders (PBDs), in which deficiency of multiple functions are caused by the absence or abnormality of peroxisomes, and disorders with isolated enzyme deficiencies in which the structure of the peroxisome is preserved or slightly altered. With the exception of X-linked adrenoleukodystrophy (X-ALD), the inheritance pattern is autosomal recessive. 2
PBDs are classified into rhizomelic chondrodysplasia punctata (RCDP) and Zellweger spectrum disorders (PBD-ZSD). The biochemical alterations of these disorders are: Zellweger syndrome (ZS), the most severe form of the Zellweger spectrum; neonatal adrenoleukodystrophy (NALD), the intermediate severity variant; and infantile Refsum disease (IRD), the mildest variant. Clinical manifestations vary according to the age of onset and severity. 2,3
The peroxisome assembly in mammals involves protein products or peroxins from 16 PEX genes and defects have been observed in 14 of these PBDs. 4 The estimated frequency of this type of disorder in North America is 1 case per 50 000 births, but it could be higher taking into account the introduction of neonatal screening for peroxisomal diseases. 5 The measurement of very long chain fatty acids (VLCFA) in plasma is the gold standard for the diagnosis of PBD, and most patients with PBD-ZSD have alterations in 4 parameters: C26:0, C26:1, C24:0/C22:0, and C26:0/ C22:0. 6,7 Molecular diagnosis is also available and is very useful given the heterogeneity of the manifestations. 8
PBDs are not included in neonatal screening programs; however, in some U.S. states, VLCFA levels are already being measured to detect X-ALD cases, using a combination of liquid chromatography and tandem mass spectrometry with blood drops on filter paper. This has also allowed detecting other disorders with high levels of VLCFA. 3
In the neonatal period, PBDs cause dysmorphism, feeding problems, marked hypotonia, seizures, impaired liver function and bone abnormalities 9, leading to significant mortality rates. The case of a newborn with clinical manifestations compatible with PBD is presented below.
CASE PRESENTATION
This is the case of a Caucasian female newborn from Boyaca, Colombia, who was referred to the hospital three days after her birth due to two seizures in the delivery room. She was the child of working-class parents that were not related by consanguinity and had already had a child with convulsive syndrome in the delivery room and dysmorphism who died after three days of life. Family history reports that a cousin of the baby died 3 days after his birth; it is important to clarify that the parents of this deceased child are siblings of the parents of the girl whose case is presented here (Figure 1).
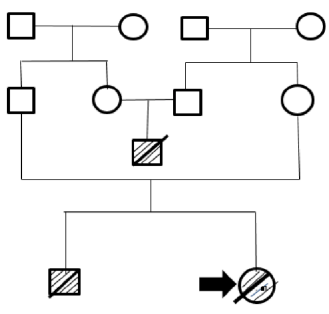
Note: the square indicates male sex and the circle, female sex. The deceased infants have a crossed line and the reported case is marked with an arrow. Source: Own elaboration.
Figure 1 Family tree.
Physical examination on admission yielded: weight: 3 700g, height: 49cm, head circumference: 37cm, thoracic circumference: 37cm, wide fontanelle, low nasal bridge, low-set ears, orbital hypertelorism, simian crease, short neck with excess skin, bilateral corneal opacity, skin jaundice, soft tissue edema and weak pulse. The neurological examination showed hypotonia, hyporeflexia and poor response to stimuli; Moro, sucking and grasp reflexes were absent.
The baby was hospitalized in the neonatal intensive care unit, where she received intravenous fluids. Laboratory tests were taken, and blood count, urinalysis, blood glucose, electrolytes, nitrogenates and aminotransferases were normal, while total bilirubin was increased with cholestatic pattern.
A computerized axial tomography of the brain was taken six days after birth, revealing ischemic lesions in white matter and cerebral edema. The following day, the condition of the patient began to deteriorate, presenting with abdominal distension and biliary drainage through the gastric tube. A simple x-ray of the abdomen showed pneumoperitoneum that required surgery; duodenal perforation was found during the procedure. Given her clinical condition, mechanical ventilation and parenteral nutrition were indicated. Subsequently, she underwent echocardiography which showed persistence of ductus arteriosus and pulmonary hypertension. The blood culture was negative.
During her hospital stay, the patient presented anemia, thrombocytopenia, prolonged coagulation times, hypoalbuminemia, hypocalcemia, conjugated hyperbilirubinemia, normal transaminases, microscopic hematuria and discrete proteinuria, as well as arterial blood gases that showed compensated respiratory acidosis. The infant suffered an episode of fever, looked septic and developed hepatosplenomegaly. A strain of Staphylococcus epidermidis was isolated by blood culture and vancomycin+amikacin was prescribed. The patient received multiple transfusions of red blood cells, platelets and plasma to treat her anemia, thrombocytopenia, prolonged clotting times and hypoalbuminemia; no adverse reactions were observed.
A cranial sonography showed grade II intracranial bleeding with discrete ventricular dilatation; metabolic screening reported negative values for qualitative tests in urine: dinitrophenylhydrazine, ferric chloride, nitroprusside and nitrosonaphthol. In addition, there were traces of reducing sugars, increase in tyrosine band in thin-layer chromatographic screening of amino acids in urine, and ammonium and lactic acid levels of 477 µmoles/L and 3.2 mMol/L, respectively. The karyotype result was 46 XX.
The patient died at 20 days of age. Necropsy reported jaundice in all organs, ascites, multiple cysts in the kidneys and dilation and bile plugs in the bile ducts (biliary dysgenesis). A peroxisomal disease was suspected due to the history of her brother, who died from a similar condition, the presence of dysmorphism along with neurological, liver and kidney involvement, and the findings of the necropsy.
Blood and urine samples were sent to the Centro de Diagnóstico de Enfermedades Moleculares (Center for the Diagnosis of Molecular Diseases) of the Universidad Autónoma de Madrid, to determine the values of pristanic, phytanic and VLCFA acids in plasma by GC/ EI-SIM-MS [gas chromatography(GC)/with electron ionization (EI) in selected ion monitoring (SIM)- mass spectrometry (MS)]. This analysis found increased VLCFA C24:0 and C26:0, ratios of C24:0/C22:0 and C26:0/C22:0, and slight elevation of phytanic acid, constituting a profile compatible with PBD (Table 1). After obtaining and analyzing these results, the family was informed on the diagnosis, risk of recurrence, and lack of treatment for the disease.
Table 1 Values of pristanic, phytanic and very long chain fatty acids in plasma and quotients.
| Analysis | Result | Reference values |
| Pristanic acid (µmol/L) | 0.33 | 0.41±0.25 |
| Phytanic acid (µmol/L) | 6.75 | 3.17±2.28 |
| C22:0 (µmol/L) | 43 | 50±16 |
| C24:0 (µmol/L) | 83 | 38±14 |
| C26:0 (µmol/L) | 26.96 | 0.55±0.17 |
| C24:0/C22:0 | 1.91 | 0.77±0.12 |
| C26:0 /C22:0 | 0.624 | 0.12±0.004 |
Source: Own elaboration.
DISCUSSION
Seizures at birth are very rare and suggest antepartum encephalopathy. 10 However, according to the study by Pisani et al. 11, in the first 28 days of life, they have a prevalence of 2.29 cases per 1 000 live births and the main causes are asphyxia at birth, brain malformations, intraventricular bleeding, meningitis and metabolic diseases.
In the case presented here, the initial manifestations (convulsion in the delivery room, dysmorphism and neurological symptoms due to hypotonia and absence of primitive reflexes) and the family history of a deceased sibling with a similar picture led to suspect a disease of genetic origin. Considering the appearance of cholestasis in the first week of life, which is a good indicator of liver disease 12, and the subsequent development of severe liver dysfunction (prolonged coagulation times, hypoalbuminemia and hyperammonemia), the possibility of an inborn error of metabolism was considered given that metabolopathies are some of the causes. 13
Some metabolic diseases may cause early seizures (pyridoxine-dependent epilepsy, peroxisomal diseases 10, non-ketotic hyperglycemia, mitochondrial diseases 11, sulfite oxidase deficiency 14, molybdenum cofactor deficiency 15, glutaric aciduria type I 16, among others) and dysmorphism (peroxisomal disorders, pyruvate dehydrogenase complex deficiency, cholesterol biosynthesis disorders, mevalonic aciduria, Smith-Lemli-Opitz syndrome, 3-hydroxyisobutyric aciduria, glutaric acidemia type II, D-2-hydroxiglutaric aciduria and mitochondrial disorders). 17 Peroxisomal and mitochondrial diseases present with dysmorphism and multiple organ involvement, and the latter are commonly accompanied by hyperlactatemia.
Dysmorphism and kidney (microscopic hematuria and proteinuria), heart (patent duc-tus arteriosus and pulmonary hypertension), liver and central nervous system involvement evidenced in the reported case pointed to a systemic disease, perhaps a peroxisomal disease resembling ZS.
The findings of this case are consistent with those presented by Zellweger 18 in a pair of siblings: hypotonia, seizures, osteotendinous areflexia, absent Moro and withdrawal reflexes, cholestatic jaundice, hypoprothrombinemia, multiple renal cortical cysts, wide forehead, low nasal bridge, wide fontanelles, low-set ears, orbital hypertelorism, simian creases, short neck with excess skin, bilateral corneal opacity, congenital heart defect and death in the first year of life.
The first step and the most widely used procedure to diagnose peroxisomal diseases is the determination of VLCFA: hexacosanic acid level (C26:0), C26:0/docosanoic acid ratio (C22/0), and tetracosanoic acid ratio (C24:0)/C22:0. VLCFA levels are elevated in PBD-ZSD and some isolated deficiencies, but they are normal in rhizomelic chondrodysplasia punctata. 6
In this case, the determination of the VLCFA showed an increase in the levels of C24 and C26 and a discrete elevation of phytanic acid, which together with the clinical findings made it possible to diagnose PBD. A level of C26>3.34 µmol/L and a C26/C22>0.10 ratio, such as those found in the reported patient, are diagnoses of ZS according to the study by Subhashini et al. 19, performed in a cohort of 90 cases and 111 controls. In addition, the presence of tyrosiluria in urine amino acid chromatography had previously been reported by Yamaguchi et al. 20 in a cohort of 20 Japanese patients with peroxisomal diseases.
Molecular studies are used to confirm the diagnosis of metabolic diseases, to establish carrier status and genotype-phenotype relationship, and to make prenatal and preimplantation diagnosis. 21 Since the phenotypic expression of peroxisomal diseases is very heterogeneous, the molecular defect definition is useful in intermediate severity forms that may not show alterations in VLCFA. 8 The present case did not have molecular studies due to the technological and economic limitations of the Colombian health system; however, the biochemical alterations were conclusive to diagnose PBD.
Although PBD-ZS has no treatment, its management should be multidisciplinary and, given the broad phenotypic spectrum, it should aim at improving the living conditions of patients, so it seeks to reduce feeding difficulties, improve liver function with supplementation of vitamin K and bile acids, manage and control seizures, and monitor sensory deficits and adrenal and kidney functions. 3 The prognosis of this disease is bleak and may lead to neonatal mortality, as in this case; however, intermediate severity cases that reach adulthood and behave as degenerative diseases may be found.
CONCLUSIONS
When dysmorphism occurs, it is necessary to establish whether it is an isolated finding and whether other organs or systems are involved. In the reported case, this disorder was accompanied by seizures in the delivery room and neurological, liver, kidney and heart involvement, manifestations that, added to the history of a deceased sibling with similar symptoms, led to the suspicion of a systemic, metabolic and genetic disease, perhaps a peroxisomal disease that resembled ZS.
Inborn errors of metabolism should be included in the diagnosis of dysmorphic neonates, especially those with multisystemic involvement. In this sense, the judicious elaboration of the clinical history and the support of laboratory tests and imaging reports are essential to propose a possible diagnosis.
Colombia is far from having a screening program for peroxisomal diseases, since the introduction of an expanded screening similar to that of developed countries is still under discussion. Therefore, it is highly relevant to establish a diagnosis, as in the case of this report, to be able to give advice to the family.

