











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCTION
Ataxia-telangiectasia (AT), also known as Boder-Sedgwick syndrome or Louis-Bar syndrome, is a multisystemic, neurodegenerative, autosomal recessive disease associated with the ATM gene mutation (A-T-mutated), which is located on the long arm of chromosome 11 (11q22-23). 1 This syndrome affects multiple organs of the body and has moderate and severe long-term sequelae.
AT is characterized by progressive neurological dysfunction with multisystemic alterations and predisposition to cancer. The characteristic symptoms of this disease usually appear early in childhood and include cerebellar ataxia, oculomotor apraxia, chorea, and cognitive impairment. 2 Moreover, the cells of AT patients present chromosomal instability, hypersensitivity to X-rays, propensity to lymphoid neoplasms, variable immunodeficiency, and susceptibility to infections, which cause systemic symptoms such as endocrinopathies, leukemias, radiosensitivity and ocular telangiectasias. 3,4
While over 400 mutations of the ATM gene have been detected, it has been reported that AT has an incidence of 1 between 40 000 and 100 000 people 5 and that its frequency of heterozygosity in the mutant allele is 1.4% to 2% in the general population. 1,3
This disease is usually diagnosed late, as it is only identifiable when patients have severe symptoms that begin with alterations in the motor system. In addition, it is often misdiagnosed as cerebral palsy. 6
So far there is no specific treatment for AT, so its management is palliative and supportive. 7 Patients with this disease should participate in daily and social integration activities, always trying to maintain their quality of life.
CASE PRESENTATION
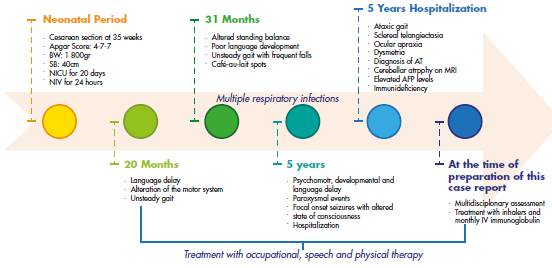
Mestizo, female patient, 7 years old, from Bogotá D.C., Colombia, and the product of a second pregnancy by young, non-blood related, middle-class parents. The girl had no significant prenatal history and was born by cesarean section at 35 weeks of gestation. Her weight at birth was 1 800gr and her size was 40cm; the APGAR score was 4/1 0, 7/10 and 7/10 at 1 minute, 5 minutes and 10 minutes, respectively. She also presented with neonatal hypoxia, requiring mechanical ventilation for 24 hours and hospitalization in the neonatal intensive care unit for 20 days.
The child's psychomotor development was normal until the age of 20 months, at which time it was evident that there was language delay, café-aulait spots and unsteady gait. At 31 months, she was referred to the Neuropediatrics Service, which requested magnification and brain magnetic resonance imaging (MRI) studies; management with physical and language therapy was initiated.
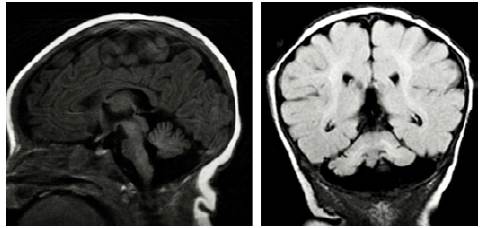
At 5 years of age, the patient was taken to the emergency department due to a possible focal onset impaired awareness seizure. During the interview, psychomotor and language delays were observed, as well as multiple respiratory and gastrointestinal tract infections. The neurological examination revealed bradylalia, bradypsychia, ataxic gait with enlargement of the support polygon, presence of oculomotor apraxia, dysmetria and telangiectasias in sclera (Figure 1), so more specific tests were requested. Finally, since MRI showed cerebellar atrophy (Figure 2), video-electroencephalography (EGG) was normal, alpha-fetoprotein (AFP) levels were elevated, and immunological tests were altered (Table 1), a diagnosis of AT was suggested.
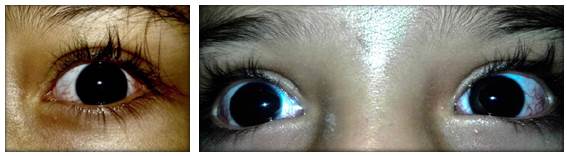
Source: Document obtained during the study.
Figure 2 Brain MRI showing cerebellar vermian atrophy. A) Sagittal plane / T1 sequence; B) Coronal plane / FLAIR sequence.
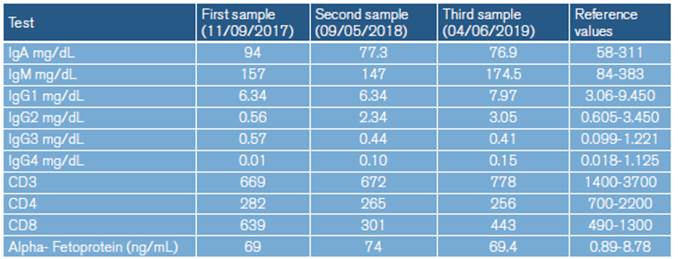
The child was assessed by the Pediatric Immunology Service, which considered cellular and humoral immunodeficiency (IgG2 and IgG4 deficiency), so she started treatment with gamma globulin IV every 28 days. She was also assessed by the pediatric hematology-oncology service due to bicytopenia (leukopenia+thrombocytopenia) during a hospital stay without evidence of neoplastic alteration. The patient continued attending child psychiatry and physical, occupational and language therapy.
The specialties of pediatric neurology and clinical genetics gathered and concluded that it was not necessary to perform a genetic study since AT could be diagnosed due to the elevated AFP levels, the evolution of the symptoms, the findings on physical examination and the associated comorbidities.
At the time of preparation of this case report, the patient had undergone a global evaluation of the disease (AFP levels, immunological tests, video telemetry and blood counts) which showed, on the one hand, an adequate response to the treatment and, on the other, the absence of new alterations or comorbidities. It is worth mentioning that, despite the poor and uncertain prognosis that AT usually has, the child did not present significant clinical deterioration and had adequate school performance, good participation in daily activities and adequate social integration. Furthermore, the number of hospitalizations due to infections and comorbidities associated with basic pathology decreased (Figure 3).
DISCUSSION
AT is a progressive, rare, autosomal recessive neurodegenerative disorder consisting of a multisystemic disorder affecting especially the neurological and immunological areas.
According to Delfino etal.,8 AT was first described in 1926 by Syllaba and Henner, who observed progressive choreoathetosis and ocular telangiectasia in three family members. Later, in 1941, Louis-Bar 9 reported progressive cerebellar ataxia and cutaneous telangiectasias in a Belgian boy, but it was not until 1957 that Boder, Sedwick and Biedmond described it as a distinct clinical entity characterized by abnormalities of organic development, neurological disorders and recurrent pulmonary infections. 10,11
The product of the ATM gene is a protein associated to the DNA repair process and the cell cycle, and it has multiple mechanisms of action. These include the phosphorylation of different substances such as the p53 tumor suppressor protein, which is responsible for stopping the cell cycle or apoptosis; 12 the protein tyrosine kinase c-Abl, involved in the repair of DNA after ionizing radiation; 13 the tumor suppressor BRCA1, involved in breast cancer 11,13-15; and the protein phosphatase 2A, which regulates the nuclear import of histone deacetylase 4 (HDAC4) and neuronal gene expression. 16,17
Abnormalities in ATM-mediated functions damage DNA by poor cell repair, accumulation of somatic mutations, and increased sensitivity to ionizing radiation, and this may cause increased risk of cancer, early aging, and neurodegeneration. 6 Similarly, thymocytes, immature B-lymphocytes, Purkinje cells of the cerebellum, and vascular endothelium are compromised by the presence of these abnormalities. 1,8
The clinical presentation of AT usually begins with progressive cerebellar ataxia associated with deterioration of fine and gross motor skills, oculomotor apraxia, nystagmus, and delayed onset and speed of speech. 18 Telangiectasias appear between the ages of three and five, mainly in the bulbar conjunctiva, nose, face, neck, and palate veil, as well as both humoral and cellular immunodeficiency (in approximately 70% of the patients). 19 Associated systemic findings include lung disease, radiation sensitivity, neurodevelopmental delay, skin conditions (hypertrichosis, seborrheic dermatitis, vitiligo, acanthosis nigricans), insulin-resistant diabetes mellitus and increased incidence of malignancy. 5
AT can appear in three different forms: Pure A-T, where patients have all or almost all of the diagnostic symptoms; attenuated or type II T-A, where patients lack some of the typical findings but have radio sensitivity; and carrier T-A, where patients with a single ATM gene mutation are at increased risk of developing cancer. 20
Regarding diagnostic criteria, Delfino et al.8 and Lazo-Rivera & Pastor-Vizcarra, 21 in accordance with the Lederman's group from the Ataxia-telangiectasia Clinical Center, established three characteristic neurological findings of AT: A) ataxic gait at the ages of 2-3; B) dysarthria and oculomotor disorders such as apraxia; and C) associated movement disorders, progressive ataxia, hypomimia, swallowing disorders and peripheral neuropathy. These authors, together with Cabana et al.,22 also stated that at least one of the following conditions must be met: ocular telangiectasia, elevated AFP level, and spontaneous or X-ray-induced chromosome breakage.
In this sense, the diagnosis is made based on clinical findings, identification of mutations in the ATM gene, elevated levels of AFP of at least 2 standard deviations (which was found in the case presented here with a sensitivity of 95%), humoral and/or cellular immunodeficiency and increased chromosomal breakage after exposure to radiation, besides the semiological characteristics exposed. 3
This report presents the case of a patient with classic AT phenotype, ataxic gait from an early age, oculomotor apraxia and dysarthria, who developed ocular telangiectasias at the age of 5. This allowed to suspect the AT diagnosis, which was later confirmed with the elevation in AFP levels (69 mg/dL); given the findings it was not necessary to do ATM gene sequencing. It should be noted that the only ataxia associated with elevated AFP is ataxia-telangiectasi a, a fact known for over thirty years and reported in over 95% of patients with this disease. 23
Although cellular or humoral immunodeficiency are not diagnostic criteria for AT, they may lead to this diagnosis; in this patient, it was possible to identify them because of the associated recurrent respiratory infections. Furthermore, regarding AT, the literature also reports defects in the T- and B-lymphocyte system, thymic hypoplasia, absence of IgA and IgE (in 70% and 80% of patients, respectively) and IgG deficiency (predominantly G2). 7
AT increases cancer susceptibility by increasing radiosensitivity and chromosomal instability, causing a genetic and telomeric break. 16 Therefore, although no neoplastic alterations were detected in the reported patient, the hematology-oncology service should monitor her since the early detection of oncological problems is important for the prognosis and follow-up of cancer.
Finally, it should be noted that since the population mentioned is not representative, no epidemiological data can be generated or generalized about the semiology or clinical benefit of treatment for AT.
CONCLUSIONS
The diagnosis of AT is based on clinical semiology and should be considered in patients with early-onset cerebellar ataxia, altered language development, and subsequent appearance of telangiectasias (at three to five years of age). However, the disease should be confirmed by verifying elevated levels of AFP and, if necessary, a genetic study, all in order to generate an appropriate clinical approach to better guide the management and prognosis of patients.
Although no curative therapy exists, symptomatic treatment for AT should be timely and interdisciplinary and should include physical therapy, occupational therapy, speech therapy and pediatric immunology, pediatric hematology-oncology, physiatry, nutrition, child psychiatry, and pediatric neurology services.

