











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
The OEIS complex is a rare polymorpic malformation. Its worldwide prevalence has been estimated between 0.04 and 0.05% in live newborns or 1 case per 200 000-400 000 pregnancies, with a male-to-female ratio of 1:2 1,2. According to Mallikarjunappa & Ghosh 3, the first case of this syndrome was reported in 1709 by Littre. However, as stated by Keppler-Noreuil 4, it was not until 1978 that Carey et al. described a congenital syndrome with multiple abdominal wall malformations, including omphalocele, cloacal exstrophy, imperforate anus, and spinal defects, which was called the "OEIS complex" 3,4. Other conditions associated with this disorder have also been discovered over time, including spina bifida, congenital urological anomalies, renal anomalies, pubic symphysis diastasis and limb abnormalities 5.
The spinal anomaly associated with the OEIS complex is occult spinal dysrafism 6, which is defined as a group of congenital malformations of the spine and spinal cord characterized by failure of fusion (total or partial) of neural structures, bone, and midline mesenchymal fields 7.
OEIS is considered a complex as it comprises morphological defects that share a common or adjacent embryological region. Its etiology is not yet clear, but it is believed to involve genetic and epigenetic factors 8. The degree of malformation depends on the prenatal period in which the primary defect occurs 9. Its prognosis is unfavorable, so early family management and counseling is always necessary.
The present article describes the case of an infant diagnosed with OEIS complex in order to emphasize the scarcity of information on this entity, especially in Latin America, and to inform about the treatment options available to date.
CASE PRESENTATION
This is the case of a 7-month-old female patient (at the time of writing this report). The mother came from a rural region of Colombia and this was her third pregnancy. Her parents were not related by blood and were low-income farmers. The pregnancy was at high obstetric risk due to advanced maternal age and prolonged intergenesic period (>11 years). The mother did not report exposure to toxics or psychoac-tive substances during pregnancy and had adequate prenatal check-ups. Her TORCHS profile was negative. A prenatal ultrasound scan (at 20 weeks) allowed diagnosing omphalocele associated with abdominal wall malformations. Fetal karyotyping revealed 46XX.
The patient was delivered through a cesarean section at 34 weeks gestation due to preterm labor caused by pre-eclampsia with severity criteria: APGAR 5/10 per minute, 7/10 at 5 minutes, and 8/1 0 at 10 minutes. At birth, her weight was 2 215g; head circumference was 32cm; height was 43.5cm. Induration in the lumbosacral region, bladder exstrophy due to a defect in the midline of the abdominal wall, bilateral talipes equinovarus and cloacal malformation were observed. Therefore, a possible OEIS complex was considered and other differential diagnoses such as gastroschisis, limb-body wall complex and pentalogy of Cantrell were ruled out.
On the third day of life, the baby was taken to skin vesicostomy, omphalocele closure, tubularization of the colonic pouch and intestinal bypass, with favorable postoperative evolution. During her hospital stay, a cranial ultrasound was performed, finding no alterations. A urinary tract ultrasound also showed grade 4 hydronephrosis, necessitating a right nephrostomy. Two days later, the infant was assessed by the pediatric neurology and neurosurgery services, which established that she presented with hypotonia and a lumbosacral mass of approximately 4x4cm, paraparesis, and hyporeflexia of lower limbs and talipes equinovarus. As a result, MRI of the neuraxis was performed, revealing lumbosacral lipomeningocele, type II diastematomyelia (Figure 1), and sacral agenesis. Occult spinal dysrafism was consideredthat did not require immediate intervention was considered. However, this condition had to be followed up on an outpatient basis and treated through rehabilitation with physical and occupational therapy.
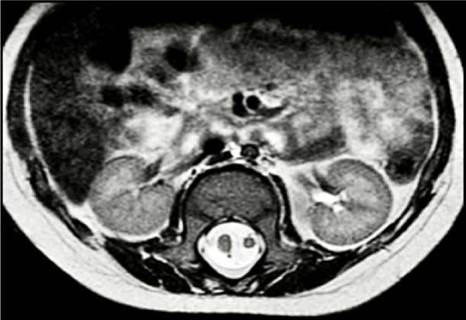
Source: Document obtained during the course of the study.
Figure 1 Magnetic resonance imaging. Axial plane. T2 sequence with evidence of type II diastematomyelia, single dural sac, and spinal cord with no bony septum/spur.
At 6 months, the patient was taken back to surgery for a bilateral pelvic osteotomy and colostomy remodeling by the pediatric orthopedic and pediatric surgery services. These procedures allowed for proper positioning and bone consolidation in the pelvis and did not cause further complications.
At the time of writing the present case report, the patient, aged 7 months, did not present any significant clinical deterioration, had adequate response to the treatment provided to alleviate her symptoms, and did not develop any postoperative complications despite the uncertainty of her prognosis. Furthermore, she was awaiting spinal anchoring and interdisciplinary follow-up by the pediatric neurology, neurosurgery, urology, pediatric surgery, interventional radiology, and physical and occupational therapy services. Clinical genetics requested genomic hybridization + microarray to identify any pathogenic variant, deletion or copy number variation that could explain the etiology, but no report has yet been received.
DISCUSSION
The first case of a patient with an OEIS complex was reported in 1709. However, Carey et al. characterized this entity for the first time in 1978 4,9,10 after identifying 175 children with one or more of the following malformations after reviewing the medical records from a California hospital: omphalocele, cloacal exstrophy, imperforated anus, and spinal defects. According to Austin et al.11, Meizner was the first to perform an ultrasound diagnosis of the OEIS complex in 1985.
OEIS is defined as a complex since it comprises a group of defects that share an embryological region and stage. It occurs at the end of blastogenesis (fourth gestational week) 12, when important embryological processes occur, such as closure of the neural tube, transverse and longitudinal folding of the embryo (formation of the anterior chest and abdominal wall), development of the midline and laterality, disappearance of the chorionic cavity due to the expansion of the amniotic cavity, formation of the umbilical cord and the cardiovascular system, onset of kidney development and initial growth and patterning of the limbs 13,14, explaining the characteristic polymorphic malformations of the entity.
Concerning pathogenesis, there are several theories that postulate four major defects: 1) failure in the formation of the urorectal septum, which prevents the separation of the urogenital and anorectal tract; 2) total rupture of the cloacal membrane and failed junction of genital tubercles and pubis branches; 3) alteration of ventral abdominal wall closure secondary to abnormal lateral folding, and 4) incomplete development of the lumbosacral vertebrae and failure of cranial neural tube closure 15,16. The reported patient showed phenotypic effects associated with the above-mentioned mechanisms. The first caused persistent cloaca, imperforate anus and genitourinary alterations such as hydronephrosis; the second caused cloacal exstrophy; the third led to an abdominal wall defect, and the fourth led to occult spinal dysraphism with diastematomyelia and tethered cord syndrome, as well as sacral agenesis.
Most OEIS complex cases are isolated and caused by a multifactorial alteration involving environmental factors such as smoking and exposure to benzodiazepines during pregnancy 17 and genetic factors such as deletion of 9q 34.1-q, 1p36 and 3q12.2-3q I1.2, trisomy 18, mosaic Turner syndrome, mutations in mitochondrial 125rRNA and mutations in homeobox genes such as HLXB9 since recurrence has been reported in some families with monozygotic conjoined twins 2,18-20. Almost all cases are premature with low birth weight; however, there are reports of patients with an average gestational age of 37.5 weeks and proper weight, height, and head circumference for age 21.
Cohen 22 suggests that the proximity of the neural tube to the cloaca during embryonic development may explain cloacal abnormalities related to occult spinal dysrafism, an entity that includes a broad spectrum of congenital fusion abnormalities of one or more dorsal midline structures. These abnormalities can affect the skin, subcutaneous tissue, vertebral bodies, meninges, and neural tissue.
There are two categories of occult spinal dysrafism: open spinal dysraphism, associated with skin defects and exposed neural tissue, and the closed spinal dysraphism, characterized by subcutaneous masses (Figure 2).
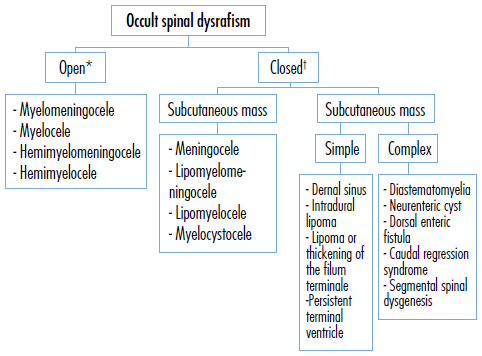
* It accounts for 98% of all cases of spinal dysrafism. † It accounts for 2% of all cases of spinal dysrafism. Source: Own elaboration based on Copp et al.23 and Wallingford et al.24.
Figure 2 Classification of occult spinal dysrafism
Although myelocystocele is the most common spinal dysrafism in patients with OEIS 25, the reported patient had a lipomeningocele, which is a herniation of cerebrospinal fluid, meninges, and neural tissue through the posterior spinal bone defect. It is also associated with the presence of lipomas that extend from the subcutaneous cell tissue to the spinal canal and with diastematomyelia, which is characterized by a "splitting" of the spinal cord in one or more segments. According to Tortori-Donati et al.26, it is classified into two types:
Type I: The arachnoid mater and the dura mater do not divide. There is only one dural sac for both hemicords. It represents 60% of diastematomyelias and 50% occur without bony spur.
Type II: The arachnoid mater and the dura mater divide into two and contain both hemicords, so each has its own subarachnoid space that joins up and down forming a single subarachnoid space. 95-100% occur with bony spur 27.28.
Alterations associated with the OEIS complex include cardiac anomalies; kidney anomalies, as in the patient reported here, who presented with grade 4 right ureterohydronephrosis that required percutaneous nephrostomy, increased nuchal translucency, and elevated serum alpha-fetoprotein 29,30.
For prenatal diagnosis, several ultrasound criteria have been classified as major and minor 11 (Table 2). Nevertheless, despite the existence of such criteria, not all anomalies can be identified prenatally and are often mistaken for some differential diagnoses such as omphalocele or gastroschisis. Therefore, the diagnosis is usually confirmed with other imaging aids such as magnetic resonance or color Doppler, used to ratify bladder exstrophy and differentiate it from the omphalocele 10,31.
Table 2 Major and minor criteria for prenatal ultrasound diagnosis of OEIS complex.
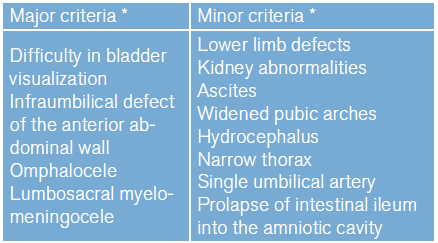
*Observed in more than 50% of patients.
Source: Own elaboration based on Copp et al.33
Patients with OEIS complex require immediate multidisciplinary care, followed by individualized surgical treatment to close the abdominal wall safely, prevent short bowel syndrome and urinary and fecal incontinence, preserve kidney function, and achieve functional and aesthetic genital reconstruction. While most cases are sporadic, family cases have been reported, which could suggest one or more specific genes that may play a key role. This makes genetic counseling and study of the utmost importance 8,34.
The prognosis of patients with OEIS complex varies depending on the severity of structural defects, the extent of cloacal exstrophy (due to its renal and pulmonary complications), and the severity of the neural tube defect. Thus, an adequate interdisciplinary management of the less severe forms may improve prognosis and lethality of this entity.
The patient in the case described here had the advantage that her OEIS complex was not lethal and that she was treated in a hospital with various medical specialties where she could undergo diagnostic imaging and receive the required medications, which allowed for a more comprehensive diagnosis-treatment with greater benefits. However, it should be noted that at the time of the writing this article the patient had not undergone all the procedures indicated since this is an entity that requires life-long interventions, management, and surveillance.
CONCLUSIONS
The OEIS complex is a rare and complicated congenital condition that can only be detected in less than 25% of cases by ultrasound in the second trimester of gestation due to the wide spectrum of anatomical variants. This occurs because malformations depend on the degree of cloacal septation and, therefore, other imaging resources such as MRI or color Doppler are often required to confirm the diagnosis.
The prognosis of the OEIS complex is unfavorable, so an interdisciplinary team of neonatologists, geneticists, pediatricians, urologists, pediatric surgeons, neurosurgeons, orthopedists, pediatric neurologists, radiologists and maternal-fetal specialists is needed to provide parents with comprehensive counseling to define pre-birth management, plan appropriate perinatal management, and achieve a better quality of life for patients. For this reason, it is important to raise awareness of this entity since an adequate clinical approach will help to better guide treatment and prognosis.

