











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroduction
Hemolytic Uremic Syndrome (HUS), first described by Gasser et al. in Switzerland in 1955, is defined by the triad of autoimmune hemolytic anemia, thrombocytopenia and acute kidney failure, in which underlying injuries are mediated by the onset of TMA. HUS is a disease that affects primarily renal vessels, but it may also affect other organs when its critical phase is triggered after the prodromal period. This phase includes symptoms such as arterial hypertension, acute kidney failure, hematological changes and extrarenal compromise that may involve heart, brain, pancreas and liver[1,6].
Traditionally, HUS is divided into two forms. The most frequent one (90% of cases) is called typical or regular HUS. It is associated with an intestinal infection caused by either Shiga toxin-producing Escherichia coli (STEC) or Verotoxin-producing Escherichia coli (VTEC), which triggers thrombotic events by directly damaging the endothelium. Less frequently, pregnancy, certain diseases (lupus erythematosus, malignant hypertension, malignant tumors), post-transplant period and use of some drugs (oral contraceptives, cyclosporine, cisplatin, gemcitabine) have also been associated with the onset of TMA and could be considered complement amplifying conditions that unmask a real clinical picture of aHUS[3,5,7,11].
The remaining 10% of HUS cases result from disorders in the regulation of the alternative complement pathway. This HUS form is called atypical HUS (aHUS) and can be accompanied by complement amplifying conditions, as mentioned above. aHUS is a serious clinical entity, with poor prognosis and high morbimortality, as can be inferred from the fact that, during the year following diagnosis, 50% of patients die, require dialysis or develop permanent renal failure[3,8].
Endothelial damage induced by complement system, due to mutations or polymorphisms in genes codifying some modulator proteins thereof, is the critical factor that causes TMA. The deeper understanding of the complement pathway gained over the last two decades has allowed to shed light on the physiopathology of aHUS and to consider complement pathway inhibition as a reasonable therapeutic option for HUS patients[4,12,13].
It should be noted that the complement system plays a critical role not only in the defense against infections, but also in the clearance of circulating immune complexes and apoptotic bodies. The system consists of proteins and can be activated through three main pathways (classical, alternative and lectin); activation, regardless of the pathway, leads to protease complex formation. Thus, C4b2a C3 convertase (resulting from either alternative or lectin activation pathways) or C3bBb C3 convertase (produced by the alternative activation pathway), cleave the C3 component into C3a and C3b, the latter being the fraction that binds to the surface of pathogens. Both C3 convertases (C4b-2a and C3b-Bb) bind to another C3b unit to form the C5 convertase, which cleaves complement component 5. In turn, C5b, a fraction resulting from this cleavage, activates pore-forming proteins (C6, C7, C8 and C9) that together form the Membrane Attack Complex (MAC). The fractions not involved in the activation cascade (C2b, C3a, C4a) mediate many biological functions such as opsonization and anaphylatoxin activities (Figure 1) (Figure 1)[5,8,13,14].
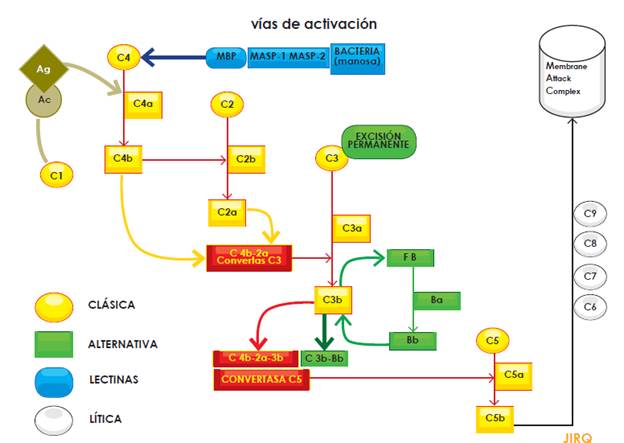
To prevent overproduction of these proteins, there is a group of regulatory proteins controlling their activation; this group includes factor H, factor I and membrane cofactor protein (MCP). These molecules maintain a low activation level in the system by regulating C3b production and locally restricting its action on the pathogen surface. Genetic or acquired alterations in these regulatory proteins are precisely those that lie beneath aHUS physiopathology; they lead to overactivation of the complement system and, in turn, to clinical manifestations of TMA due to endothelial injury. In addition, the understanding of this intricate system enabled the development of the first inhibitor of its activity: eculixumab[8,14,16].
Case Report
The patient, PJBL, was a 52-year-old man with a past medical history of polycystic kidney disease (diagnosed in 2002), stage 5 CKD in renal replacement therapy, cadaveric renal transplantation (September 11th, 2013), arterial hypertension, hypertensive heart disease (LVEF of 35%), hyperuricemia and benign prostatic hypertrophy. He was referred from Cucuta, approximately 5 months after renal transplantation because of an 8-day clinical picture including disorientation, aggressiveness, bradylalia, bradypsychia, fever, and diarrheal stools. Signs and symptoms were interpreted as a sepsis clinical picture, the cause of which was to be determined. Treatment with linezolid, ceftriaxone, ampicillin and valganciclovir (because of a potential cytomegalovirus infection) was started and routine screening as well as specialized tests were performed to exclude the possibility of neuroinfection (Table 1).
Table 1 Chronological results of tests performed on the patient
| Examen | 10 de febrero | 12 de febrero | 13 de febrero |
|---|---|---|---|
| Hemoglobin (gr/dL) gr/dL) | 14,2 | 11,9 | 11,3 |
| Hematocrite (%) | 43,1 | 33,3 | 32,0 |
| Leukocytes (cel/mm) | 10500 | 6850 | 10400 |
| Thrombocytes (cel/mm) | 95100 | 59000 | 60700 |
| Creatinine (mg/dL) | 2,5 | 2,83 | 3,23 |
| BUN (mg/dL) | 48 | 59 | 79,9 |
| Brain CT | Normal |
BUN: Blood Urea Nitrogen, CT: computed tomography.
Physical examination on admission revealed that patient was in apparent good general condition and afebrile, with incoherent language and the following vital signs: weight 80 kg; height 1.68 m; BMI 28.3; blood pressure 157/114; heart rate of 116/min; respiratory rate of 18/min; body temperature of 37 °C and O2 saturation of 86% while breathing room air. He presented conjunctival pallor, dry oral mucosa, tachycardic rhythmic heart sounds, normal vesicular murmur. Neurologically, the patient showed disorientation in the three spheres, drowsiness, limb tremor and positive meningeal and Kernig's signs.
At that moment, blood tests revealed hemoglobin of 11,9 g/dL, white blood cell count of 8450/mm3; neutrophilia of 92%; platelets of 63000/mm3. A metabolic panel showed BUN of 81.4 mg/dL, creatinine of 3.16 mg/dL (0.67 to 1.17 mg/dL) and lactate dehydrogenase of 425 IU/L (135 to 214 IU/L).
Nephrological assessment (involving the transplant team) found neurological impairment in the patient and deterioration of his general condition, with high suspicion of neuroinfection and elevation of nitrogen compounds. For this reason, transfer to the ICU was requested and immunosuppressive therapy was suspended.
After neurological evaluation, a simple cerebral computerized axial tomography was requested. Findings revealed moderate changes in cortical and subcortical atrophy related to age. A lumbar puncture revealed clear, colorless cerebrospinal fluid; glucose content of 122 mg/dL; protein level of 49 mg/dL; and chloride content of 130 mEq/L. Neither bacteria nor yeasts were observed. VDRL was non-reactive and nigrosin staining did not revealed any microorganisms. Results obtained from latex-cryptococcal antigen test were negative.
Considering these findings, brain magnetic resonance was requested. Results showed cortical hyperintensities predominantly in the occipital lobe (that could be associated with cerebritis regions); nodular image in left cerebellum hemisphere, in which a potential injury was to be considered, induced by either a tumor or, less likely, an infection (toxoplasmosis, nocardiosis, pyogenic brain abscess). For this reason, along with the infectiology team, it was decided to continue treatment with ceftriaxone, trimethoprim/sulfamethoxazole and metronidazole, considering the risk-benefit ratio.
However, given that these findings excluded an infectious process, neurologists established tacro-limus-induced encephalopathy as a probable diagnosis and suggested that a change in drug regimen should be considered once immunosuppressive treatment was resumed.
Since clinical features evolved into a worsening of renal function due to increased serum creatinine despite normal diuresis (urine output of 1.3 cc/ kg/hour), hemodialysis was started. As a result, the patient conditions improved and he was transferred to hospital ward to continue medical treatment. The nephrology unit decided to resume immunosuppressive treatment with sirolimus, mycophenolic acid and prednisolone, in low doses.
During hospitalization, detection of schistocytes in peripheral blood smear revealed progressive anemia; this led to clinical suspicion of a thrombotic microangiopathy (TMA). Results of both hemo-cultures and stool tests were negative. Cytomegalovirus viral load was undetectable and tacrolimus serum levels were 1 ng/mL (normal range from 5 to 20 ng/mL).
Then, TTP/aHUS was deemed as a plausible diagnosis because of the following findings: a) hemolytic anemia, b) moderate to severe thrombocytopenia since the start of the clinical picture and without an identified cause; c) schistocyte presence in peripheral blood; d) graft dysfunction that initially appears as acute renal injury with unknown precipitating factor; e) unknown-cause encephalopathy that diminished after hemodialysis; f) normal levels of tacrolimus, and g) early thrombosis associated with hemodialysis catheter.
To exclude other factors causing kidney failure, and considering that similar clinical pictures can stem from humoral rejection, a renal biopsy was performed. As tests revealed alterations within clotting system, fresh plasma infusion was conducted, which led to a significant improvement in the patient's medical condition (which additionally supports aHUS diagnosis). The brain magnetic resonance was reviewed by the radiological unit and interpreted to be posterior reversible encephalopathy syndrome, of which TTP/HUS is a possible cause.
Therefore, ADAMTS-13 activity was evaluated, yielding a result higher than 5% (patient's result 100%). Although testing for Shiga toxin could not be performed, clinical evolution did not suggest enteroinvasive E coli infection; in addition, up to 30% of aHUS patients present diarrhea. The renal biopsy did not indicate thrombosis and both humoral and cellular rejection were excluded as cause of kidney deterioration.
Thereafter, the medical condition of the patient continued worsening due to the development of nephrotic-range proteinuria (4.2 g in 24 hours) and increase in the number of nitrogen compounds, with persistent signs of mild TMA. Therefore, it was considered that the patient's clinical picture was compatible with aHUS unmasked using calcineurin inhibitor (tacrolimus), which explains his torpid evolution despite having suspended the medication at least two weeks before.
Given the final diagnosis of post-transplant aHUS, treatment with eculizumab was started on August 6th, according to the regime approved for this clinical entity. Complying with the administration protocol, and to avoid possible infections by encapsulated pathogens, the patient was vaccinated against meningococcus two weeks before treatment start. Since the patient had already been vaccinated against pneumococcus two years ago, this vaccine was not administered.
Hitherto (January 2016), patient is in good general medical condition. Sirolimus was replaced by belatacept (Nulojix) because of nephrotic-range proteinuria. mTOR-targeted drugs were not considered because they would have increased proteinuria. Patient presents creatinine levels between 1.7 and 2.0 mg/dL and decreasing proteinuria values (1858 mg/24 h in November 2015). He continues on humanized monoclonal antibody treatment without evidence of new TMA episodes nor renal deterioration. Finally, he recovered and significantly improved his life quality in general terms.
Discussion
aHUS is a clinical condition that, besides uncommon, is complex, polygenic and multifactorial. Hitherto, only approximately 1000 cases have been reported worldwide; for this reason, despite being estimated to 1-2 cases per million, or up to 3.3 according to recent studies, its actual incidence remains unknown. Although this condition affects primarily children and young people, it may appear at any stage of life, with an even gender distribution. Unlike HUS, which most of the times occurs as a single event with complete recovery, aHUS physiopathology determines its chronicity, multiple relapses, and poor prognosis if inadequate treatment is administered[16,17.
The onset of signs and symptoms is generally abrupt, because the classical triad (hemolytic anemia, thrombocytopenia and kidney failure) can be accompanied by an increase in blood pressure values, clinical manifestations of other affected organs (central nervous system, heart, lungs and pancreas) and proteinuria, hematuria, increased lactate dehydrogenase levels, very low haptoglobin levels and presence of schistocytes[3,4].
Among the extrarenal affections, the most frequent one is neurological compromise, manifestations of which include irritability, drowsiness, disorientation, encephalopathy, seizures and even brain stroke[17].
Nowadays, the understanding of aHUS physio-pathology is much greater than before. The complement system performs diverse functions and its activation, regardless of the pathway (classical, alternative or lectin), leads to the assembly of complexes with convertase activity. To avoid its total consumption by activation, there are complement regulatory proteins (factor H, factor I and MCP) that dissociate the convertase[14].
Penetrance in patients carrying aHUS mutations is approximately 50% (genetic polymorphism); hence, only some carriers develop the disease. Given that clinical manifestations are variable, there must be additional factors (genetic or environmental) modulating expression of the disease. Some mutations (for instance, factor H mutations) are associated with a worse prognosis than others, given that they are related to a faster progression towards chronic kidney disease, poor response to previous palliative treatments (such as plasmapheresis), and higher rates of relapses and mortality[18,19].
Several mutations, hereditary and sporadic, have been found to be associated with aHUS, such those in factor H, factor I, factor B, MCP and C3 genes. Factor H mutation is the most frequent and predisposes patients to complement system activation on the surface of their cells. This loss of regulation may arise from protein function loss, due to mutations in factor H, factor I and MCP, as well as from higher activation of C3 convertase because of alterations in factor B and C3[17,18].
Moreover, the problem of some aHUS patients is that they produce anti-factor-H autoantibodies. This could explain the onset of the condition in patients without genetic mutations; therefore, seeking this type of antibodies would be indicated in this group of patients. The possibility that there are additional antibodies with specificity against other complement system proteins is being currently addressed by other studies, as well as the involvement in aHUS physiopathology of other complement genes and proteins[5].
It is estimated that 40-60% of patients with aHUS carry heterozygous point mutations in complement system genes (especially factor H and factor I), but their penetrance is approximately 50%, which explains that only some carriers develop the disease. This, along with the fact that up to 10% of patients carry more than one mutation and that genetic variant modulate penetrance, supports the multiple-hit hypothesis for the development of aHUS[19].
De novo aHUS diagnosis in renal-transplant patients occurs in 1-5 % of the cases, especially in the three first months after transplantation. Use of anti-calcineurinics, some viral infections, acute vascular rejection after cadaveric kidney transplantation and the presence of antiphospholipid antibodies are considered aHUS risk factors[7].
In those cases, in which the onset of TMA has been associated with anticalcineurinics, diverse strategies have been used (dose reduction or anticalcineurinic replacement) and final decision should be made on an individual basis. What seems to be clearer is the improvement in TMA once calcineurin inhibitor has been replaced or its dose has been lowered. This suggests that de novo aHUS diagnosis after transplantation should be considered when TMA persists after drug suspension, as in the case of the patient in this report. Plasma infusion can be used until improvement in signs and symptoms; however, this has not shown to change the natural history of the disease and the prognosis in terms of morbimortality remains discouraging4. Also, infusion and therapeutical exchange of plasma have shown to lead to a transient hematological improvement, without major impact on kidneys or other compromised organs, as it occurred in the case reported herein. In patients with terminal renal disease who have been transplanted and have developed aHUS, the disease recurrence factor depends on which mutation they carry, considering that failure to identify it does not exclude diagnosis nor the risk of recurrence in the post-transplant period[4].
In summary, aHUS arises from genetic and environmental factors; and inadequate complement system regulation may arise from loss-of-function (factor H and factor I) or gain-of-function mutations (factor B or protein C3) or production of certain antibodies (anti-factor H), among others already described. In this scenario of dysregulation, complement system activity cannot be controlled once triggered (for instance, by pathogens, drugs, pregnancy, tumor cells, etc.) and directly induces blood clot formation by damaging endothelial cells.
Diagnostic suspicion of aHUS starts with a clinical picture of TMA. Since signs and symptoms do not allow to conduct differential diagnosis, it is necessary to perform complementary tests to exclude other TMAs and confirm clinical diagnosis of aHUS through a process of elimination. For this purpose, ADAMTS-13 activity assay and Shiga toxin test are indicated to exclude, respectively, TTP and those cases with a compatible clinical picture. Patient is diagnosed with STEC HUS if Shiga toxin result is positive; TTP if ADAMTS-13 activity is <5%; and aHUS in case that ADAMTS-13 activity is >5% and Shiga toxin test result is negative[3,4,9].
It is important to determine which mutations are involved in aHUS, as prognosis varies according to the type of mutation, even though it is still debated in the current literature. It has been found that alterations in factor H are associated with the worst prognosis. Likewise, MCP mutations that seem to have a better prognosis also have a high impact on morbimortality in the long term, which may be related to their association with other mutations. DNA profiling is especially relevant in patients with terminal kidney disease who are candidates for transplant, as well as the evaluation of both the risk of relapse and the medical strategy to be used after organ transplantation.
Despite the lack of clinical trials, plasma therapy (infusion and exchange) has been the empirical treatment of choice for patients with aHUS. Indeed, the medical condition of the patient in this report significantly improved after having received plasma to treat blood clotting alterations before he underwent kidney biopsy[9,14] Notwithstanding, as presented above, global prognosis, in terms of recurrence, morbidity and mortality, is not affected by this type of therapy.
The most recent therapy for these patients is eculizumab. This drug is a humanized monoclonal antibody that binds to complement C5 with high affinity and inhibits its cleavage to C5a and C5b. Low levels of C5a reduce the number of inflammatory cells and, more importantly, low C5b availability decreases the formation of membrane attack complex (C5b-9)[15,20].
The efficacy and safety of eculizumab have been tested and, fortunately, the response to this drug has been uniform irrespective of the level of the disease, because the drug acts blocking C5 and, thus, inhibits production of the C5 convertase and the formation of the membrane attack complex, which is partly responsible for endothelial damage and onset of TMA. This indicates that eculizumab preserves earlier functions in complement system cascade, which are crucial for microorganisms opsonization and clearance of residual apoptotic bodies and circulating immune complexes as well. Early use of eculizumab has proven not only to be more successful than plasma therapy, but also to favor kidney function recovery, even if the patient is on hemodialysis16, 20-22. Our patient showed a remarkable improvement of kidney function and quality of life after having started anti-complement treatment.
It is also important to mention that patients with a constitutional C5 deficiency have a higher risk of contracting meningitis caused by Neisseria. The use of eculizumab has been associated with this infection (approximately 4 cases out of 1,000 patients/ year) and, therefore, vaccination is mandatory in patients before its administration[22,24].
There is a relation between the severity of the acute phase and long-term prognosis. Prognosis is poorer in patients with an anuria longer than 15 days, extrarenal (mainly neurological) manifestations, or mutations in factor H and C3 genes. In contrast, regarding history of diarrhea, presence of Shiga toxin improves prognosis[17,25,26].
Conclusion
The case of a patient with renal transplant who was admitted to the hospital due to neurological manifestations is presented. After a prolonged and exhaustive study, his clinical manifestations along with laboratory findings, compatible with a TMA that did not improve upon suspending calcineurin inhibitor, and did not correspond to other likely TMA causes such as TTP either, as well as the absence of evidence of renal transplant rejection in biopsy, allowed to conclude that the patient was developing de novo aHUS in the post-transplant period.
Although these are rare pathologies, the main medical barrier is the lack of knowledge. This case allows to illustrate the clinical exercise to reach a definitive diagnosis. Any patient with the triad of hemolytic anemia, thrombocytopenia and renal failure should complete the whole exhaustive diagnostic exercise of TMA to arrive at a correct diagnosis and define the appropriate therapy.

