










Servicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Nefrología
versión On-line ISSN 2500-5006
Rev. colom. nefrol. vol.4 no.1 Bogotá ene./jun. 2017
https://doi.org/10.22265/acnef.4.1.267
Artículo casos clinicos
Síndrome hemolítico urémico atípico posterior a trasplante renal: presentación de un caso y revisión de la literatura
6Médico internista, profesor asociado, posgrado Medicina Interna, Universidad Autónoma de Bucaramanga; coordinador Departamento de Medicina Interna, Fundación Oftalmológica de Santander, Clínica Carlos Ardila Lülle, Santander, Colombia
7Médico residente, Medicina Interna, Universidad Autónoma de Bucaramanga, Colombia
8 Médico, Departamento de Nefrología, Fundación Oftalmológica de Santander Clínica Carlos Ardila Lülle, Santander, Colombia
9Médico internista, nefrólogo, Fundación Oftalmológica de Santander Clínica Carlos Ardila Lülle, Santander, Colombia
10Médico internista, nefrólogo; Magister Administración en Salud; director médico Alexion Pharma Colombia, Bogotá, Colombia
El síndrome hemolítico urémico (SHU) es una entidad clínica caracterizada por la aparición de anemia hemolítica no inmune, trombocitopenia e insuficiencia renal aguda. Se trata de una enfermedad perteneciente al grupo de las microangiopatías trombóticas (MAT) de la que hacen parte también la purpura trombocitopénica trombótica (PTT) y algunas otras MAT asociadas a otras condiciones médicas antes conocidas como MAT secundarias.
Por otra parte, la variedad conocida como SHU atípico (SHUa) es una patología ultra-huérfana que frecuentemente evoluciona a insuficiencia renal crónica (IRC) y se asocia con elevada morbi-mortalidad si no recibe el tratamiento adecuado.
Se examina el caso de un paciente que presenta su primera manifestación clínica de síndrome hemolítico urémico atípico después de trasplante renal cadavérico lo cual no solo lo hace un caso aún más exótico, sino que implica mayor complejidad en su manejo.
Palabras clave: síndrome hemolítico urémico atípico; trasplante renal; plasmaféresis; tacrolimus; microangiopatía trombótica; eculizumab
Haemolytic uremic syndrome (HUS) is a clinical entity characterized by the appearance of non-immune hemolytic anemia, thrombocytopenia and acute renal failure. It is a disease belonging to the group of thrombotic microangiopathy (MAT) which are part of thrombotic thrombocytopenic purpura also (PTT) and some other MAT associated with other medical conditions formerly known as secondary MAT. Moreover, the variety known as atypical HUS (aHUS) is an ultra-orphan disease that frequently progresses to chronic renal failure (CRF) and is associated with high morbidity and mortality if not properly treated.
If a patient presents its first clinical manifestation of aHUS later receive a cadaveric renal transplant which not only makes it an even more exotic case but involves more complexity in their management is presented.
Key words: Atypical hemolitic uremic síndrome; kidney transplant; plasmapheresis; tacrolimus; thrombotic microangiopathy; eculizumab
Introducción
El SHU, descrito por primera vez por Gasser y col. en Suiza en 1955, está definido por la triada de anemia hemolítica autoinmune, trombocitopenia e insuficiencia renal aguda, en las que las lesiones subyacentes están mediadas por la aparición de MAT. Se trata de una enfermedad que afecta predominantemente los vasos renales pero que puede afectar otros órganos de la economía, pues pasado el período prodrómico se desencadena la fase crítica. Esta incluye, entre sus manifestaciones, hipertensión arterial, insuficiencia renal aguda, cambios hematológicos y compromiso extra renal que puede incluir corazón, cerebro, páncreas e hígado[1,6].
Tradicionalmente se distinguen dos formas de SHU. La forma más frecuente (90% de los casos) obedece al denominado SHU clásico o típico. Este se asocia al antecedente de una infección intestinal causada por Escherichia coli productora de Shiga-toxina o STEC (Shiga Toxin Escherichia coli) o por otro tipo de bacterias productoras de vero-toxina (VTEC) que, ejerciendo un efecto lesivo directo sobre el endotelio, desencadenan los fenómenos trombóticos. Menos frecuentemente, situaciones como embarazo, ciertas enfermedades (lupus eritematoso, hipertensión maligna, tumores malignos), pacientes postrasplantados y el uso de ciertos fármacos (anticonceptivos orales, ciclosporina, cisplatino, gemcitabina). También se han visto asociadas con la aparición de MAT y podrían considerarse condiciones amplificadoras del complemento que desenmascaran un cuadro real de SHUa[3,5,7,11].
El 10% restante de los casos de SHU es consecuencia de alteraciones en la regulación de la vía alternativa del complemento, al cual se le denomina SHU atípico (SHUa). Este, como se mencionó previamente, puede estar acompañado de otras condiciones amplificadoras del complemento. El SHUa se trata de una entidad grave, de mal pronóstico y elevada morbimortalidad, pues en el siguiente año al diagnóstico más del 50% de los pacientes fallecen, requieren diálisis o presentan daño renal permanente[3,8].
El daño endotelial inducido por el complemento, debido a mutaciones o polimorfismos de los genes que codifican ciertas proteínas moduladoras de este sistema, es el factor crítico responsable de la generación de MAT. Un conocimiento más detallado de la vía del complemento en las últimas dos décadas ha permitido aclarar la fisiopatología del SHUa y es este entendimiento lo que ha permitido postular que su inhibición es una opción terapéutica lógica en este tipo de pacientes[4,12,13].
Debe recordarse que el sistema del complemento no solo resulta esencial en la defensa frente a las infecciones, sino que también participa en la depuración de complejos inmunes circulantes y en la eliminación de cuerpos apoptóticos. El sistema consta de una serie de proteínas con tres vías de activación (clásica, alternativa y de lectinas) en donde, sin tener en cuenta la vía, el sistema conduce a la formación de complejos proteicos con actividad de proteasa. Teniendo ello presente, la generación de la C3-convertasa tipo C4b-2a (concebida ya sea por la vía clásica o por la vía de las de lecitinas), o de la C3-convertasa tipo C3b-Bb (generada por la vía alternativa), actúa sobre el componente C3 degradándolo en C3a y C3b, siendo esta última fracción la que se adosa en la superficie del agente a eliminar. Sobre C3b se unen las dos C3-convertasas ya generadas (C4b-2a y C3b-Bb) y dan origen a la C5-convertasa. Esta proteasa degrada el factor 5 del complemento y es su fracción C5b la que desencadena la activación de las proteínas formadoras de poro en las membranas (C6, C7, C8 y C9) que en conjunto son conocidas como complejo de ataque a la membrana o MAC (Membrane Attack Complex). Las fracciones que no intervienen en la cascada de activación (C2b, C3a, C4a) tienen capacidades de anafiloxina y de opsonización (Figura 1)[5,8,13,14].
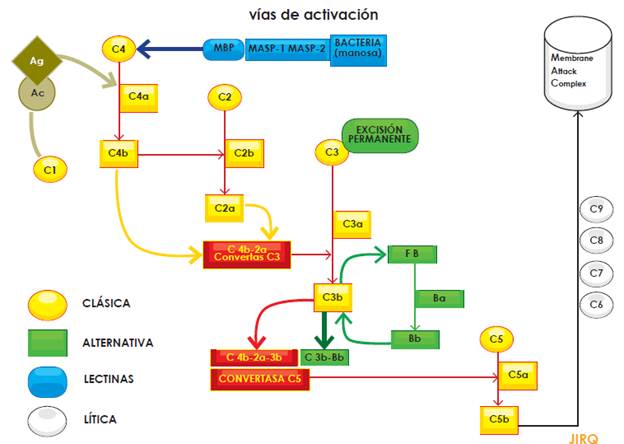
Para evitar que la generación de estas proteínas se torne excesiva, existe un conjunto de proteínas moduladoras que regulan su activación, que incluye el factor H, el factor I, la proteína cofactora de membrana (MCP). Estas moléculas, al regular la generación de C3b, mantienen un nivel bajo de activación del sistema y, si este se activa, limitan su acción localmente en la superficie del patógeno. Es, justamente en las alteraciones, genéticas o adquiridas, sobre estas proteínas moduladoras en donde radica la fisiopatología del SHUa, lo cual lleva a una sobre activación del sistema del complemento y las manifestaciónes clínicas de MAT por lesión endotelial. Además, es el entendimiento de este intrincado sistema del complemento el que permite el desarrollo del primer inhibidor de su actividad: el eculixumab[8,14,16].
Presentación del caso
Se trata de un paciente, PJBL, de 52 años de sexo masculino con antecedentes de poliquistosis renal del adulto (diagnosticada en el 2002), IRC estadio 5 en terapia de reemplazo renal (CAPD), trasplante renal de donante cadavérico (11 de septiembre de 2013), hipertensión arterial, cardiopatía hipertensiva (FEVI del 35%), hiperuricemia e hipertrofia prostática benigna. Ingresa remitido desde Cúcuta, aproximadamente 5 meses después del trasplante renal, por cuadro clínico de 8 días de evolución caracterizado por desorientación, agresividad, bradilalia, bradipsiquia, fiebre y deposiciones diarreicas. Este cuadro es interpretado como cuadro de sepsis de origen a determinar. Se inicia manejo con linezolid, ceftriaxona, ampicilina y valganciclovir (posibilidad de infección por citomegalovirus) y se solicitan exámenes de rutina, así como especializados, con el fin de descartar posibilidad de neuroinfección (tabla 1).
Tabla 1 Resultados cronólogicos de los exámenes practicados al paciente
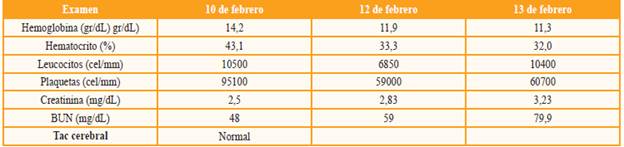
BUN: nitrógeno ureico sanguíneo, TAC: tomografía axial computarizada.
En el momento del examen físico de ingreso, paciente en regular estado general, afebril, lenguaje incoherente y con los siguientes signos vitales: peso 80 kg; talla 1,68 m; IMC 28,3; tensión arterial 157/114; frecuencia cardiaca de 116/min; frecuencia respiratoria de 18/min; temperatura de 37 °C; y saturación de O2 del 86% al ambiente. Conjuntivas pálidas, mucosa oral seca, ruidos cardiacos rítmicos taquicárdicos, murmullo vesicular normal. Neurológicamente se apreció desorientado en las 3 esferas, somnoliento, con temblor de las extremidades y con signos meníngeos y de Kernig positivos.
En ese momento los exámenes del paciente revelaron hemoglobina de 11,9 gr%; leucocitos 8450/ mm3; neutrofilia del 92%; plaquetas de 63000; BUN de 81,4 mg/dL; creatinina de 3,16 mg/dL; (0,67 a 1,17 mg/dL) y deshidrogenasa láctica de 425 UI/L (135 a 214 UI/L).
La valoración nefrológica (y por el grupo de trasplante) encontró paciente con deterioro neurológico y de sus condiciones generales, con alta sospecha de neuroinfección y con elevación de azoados. Por ello, solicita traslado de paciente a UCI y suspende el manejo inmunosupresor.
Luego de la valoración por neurología, se solicita la realización de tomografía axial computarizada cerebral simple. Esta reporta moderados cambios de atrofia cortical y subcortical relacionados con la edad. Se realiza punción lumbar obteniéndose líquido cefalorraquídeo incoloro, trasparente; glucosa 122 mg/dL; proteínas 49 mg/dL; cloro 130 mEq/L; bacterias y levaduras no se observan; VDRL no reactivo; tinta china negativo; y prueba de látex de antígeno para criptococo negativa.
Ante estos hallazgos, se solicita resonancia magnética cerebral. Esta reporta híper-intensidades corticales de predominio occipital (que podrían corresponder a zonas de cerebritis); imagen nodular a nivel del hemisferio cerebeloso izquierdo, en el cual debe considerarse posibilidad de lesión tumoral o menos probable infecciosa (toxoplasmosis, nocardiosis, absceso cerebral piógeno). Debido a esto, conjuntamente con infectología, se decide continuar el tratamiento con ceftriazona, trimetoprim-sulfametosaxol y metronidazol, evaluando la relación riesgo/beneficio.
Sin embargo, ante estos hallazgos que descartaban un proceso infeccioso, neurología determina como diagnóstico posible encefalopatía secundaria a toxicidad por tacrolimus y sugiere que, una vez se reinicie manejo inmunosupresor, se considere hacerlo con cambio de esquema.
Puesto que el paciente evoluciona con deterioro de su función renal, debido a la elevación de la creatinina sérica pese a diuresis normal (gasto urinario de 1,3 cc/kg/hora), se decide iniciar hemodiálisis. Con ello, el paciente mejora sus condiciones para ser trasladado de UCI a piso para continuar con tratamiento médico. Nefrología decide reiniciar el manejo inmunosupresor con sirolimus, micofenolato y prednisolona, en bajas dosis.
En la hospitalización se detecta anemia progresiva con presencia de esquistocitos en el extendido de sangre periférica, por lo que el diagnóstico se inclina hacia una microangiopatía trombótica (MAT). Los hemocultivos fueron reportados como negativos, coprocultivo negativo, carga viral para citomegalovirus indetectable y los niveles séricos de tacrolimus de 1 ng/ml (valor normal de 5 a 20 ng/ml).
En este momento, se contempla la posibilidad de PTT/SHUa en el paciente debido a los siguientes hallazgos: a) anemia hemolítica, b) trombocitopenia moderada a severa desde el inicio del cuadro y sin causa esclarecida, c) presencia de esquistocitos en sangre periférica, d) disfunción del injerto que debuta como lesión renal aguda sin conocimiento de causa precipitante, e) encefalopatía de causa no esclarecida que mejoró con hemodiálisis, f) niveles normales de tacrolimus, y g) trombosis temprana de sitio de catéter de hemodiálisis.
Con el fin de descartar otras causas dela falla renal del paciente, se decide la realización de biopsia renal, dado que el rechazo humoral puede presentarse con cuadros clínicos similares. Ante la presencia de alteración en las pruebas de coagulación se aplicó plasma fresco, evento que produjo una marcada mejoría en el estado clínico del paciente (y que puede tomarse como otro punto a favor de SHUa). En ese momento, una nueva revisión del servicio de radiología de la resonancia cerebral es interpretada como síndrome de encefalopatía posterior reversible, cuadro radiológico donde la PTT/SHU figura como una causa posible.
En vista de todo lo anterior, se solicitó ADAMTS-13, el cual fue mayor del 5% (resultado del paciente 100%), y no se pudo realizar test de toxina Shiga. Sin embargo, la evolución clínica no sugiere infección por E. coli enteroinvasiva y hasta el 30% de los pacientes con SHUa presentan cuadros diarreicos. La biopsia renal fue negativa para trombosis y se descartó rechazo humoral y celular como causa del deterioro renal.
Luego de ello, el paciente continuó en deterioro clínico debido al desarrollo de una proteinuria en rango nefrótico (4,2 gr en 24 horas) y elevaciones de las cifras de azoados, con persistencia de signos de MAT leve. Con lo anterior, se considera que el paciente cursa con cuadro de SHUa desenmascarado por el uso de inhibidores de calcineurina (tacrolimus), lo cual explica la evolución tórpida del paciente pese a haber suspendido el medicamento al menos dos semanas antes.
Ante el diagnostico final de SHUa pos-trasplante, se inicia manejo con eculizumab el 6 de agosto según el esquema aprobado para esta entidad. Por protocolo de aplicación y con el fin de prevenir posible infección por gérmenes encapsulados, el paciente recibió vacuna contra meningococo dos semanas previas al inicio de eculizumab y, debido a que dos años atrás había recibido vacuna contra el neumococo, esta no fue necesario aplicarla.
A la fecha (enero de 2016), el paciente se encuentra en buenas condiciones generales. Se hizo cambio de inmunosupresión de sirolimus a belatacept (Nulojix) por proteinuria en rango nefrótico. No se consideró un mTOR ya que aumenta la proteinuria. Se encuentra con rangos de creatinina entre 1,7 y 2,0 mg/dL y valores de proteinuria en disminución (1858 mg/24 h en noviembre de 2015). Continúa en manejo con Anticuerpo Monoclonal Humanizado sin evidencia de nuevos episodios de MAT ni deterioro renal. Adicionalmente, el paciente recuperó y mejoró de forma muy significativa su calidad de vida en términos generales.
Discusión
El SHUa es una entidad clínica que, además de rara, es compleja, poligénica y multifactorial. De ella, al día de hoy, solo existe el reporte de algo más de 1000 casos en el mundo, razón por la cual no se conoce su verdadera incidencia, que se ha calculado en 1 a 2 casos por millón de habitantes. Sin embargo, publicaciones recientes hablan de 3,3 casos por millón de habitantes3. Esta entidad, si bien afecta mayoritariamente a niños y adultos jóvenes, puede aparecer en cualquier época de la vida, con una distribución uniforme por género y, a diferencia del SHU que suele ser un evento único con recuperación completa la mayoría de las veces, la fisiopatología del SHUa determina su cronicidad, múltiples recaídas, y mal pronóstico en caso de no recibir el tratamiento adecuado[16,17].
El inicio del cuadro generalmente es abrupto, pues a la triada clásica (anemia hemolítica, trombocitopenia e insuficiencia renal) se pueden agregar elevación de cifras tensionales, manifestaciones clínicas indicativas de afección de ciertos órganos (sistema nervioso central, corazón, pulmón y páncreas) y alteraciones de laboratorio consistentes en proteinuria, hematuria, incremento en los niveles de deshidrogenasa láctica, niveles muy bajos de haptoglobina y presencia de esquistocitos[3,4].
Entre las afecciones extra renales, la más frecuente es el compromiso de tipo neurológico cuyas manifestaciones incluyen, entre otras, irritabilidad, somnolencia, confusión, encefalopatía, convulsiones e incluso accidente cerebrovascular[17].
La fisiopatología del SHUa está hoy muchas más esclarecida. El sistema del complemento cumple funciones variadas y, sin importar su vía de activación (clásica, alternativa o por lecitinas), todas conducen a la formación de complejos con actividad convertasa. Para evitar un consumo total por su activación, existen proteínas reguladoras del complemento (factor H, factor I y MCP) que disocian la convertasa[14].
La penetrancia en pacientes portadores de mutaciones del SHUa es de aproximadamente un 50% (polimorfismo genético), por ello solo algunos portadores desarrollan la enfermedad. La presentación clínica es variable, por lo que deben existir factores adicionales (genéticos y/o ambientales) que por modulación influyan en la expresión de la enfermedad. Algunas mutaciones se asocian a peor pronóstico pues expresan mayor progresión a enfermedad renal crónica, pobre respuesta a tratamiento paliativos previos (como la plasmaféresis), mayor tasa de recaídas y más alta mortalidad (por ejemplo, el SHUa asociado a mutación del factor H)[18,19].
Con respecto al SHUa se han descrito mutaciones, esporádicas o hereditarias, en diversas proteínas: factor H, factor I, factor B, MCP y C3, siendo la más frecuente el defecto en el factor H, cuya alteración predispone al paciente a la activación del sistema del complemento sobre las superficies celulares propias. Esta pérdida de la regulación se puede dar por dos vías, pues mientras las mutaciones del factor H, del factor I y de la MCP comprometen la función de la propia proteína, los defectos del factor B y de C3 inducen una mayor activación de la C3-convertasa[17,18].
Por otra parte, hay un grupo de pacientes con SHUa cuyo problema radica en la producción de autoanticuerpos antifactor H y que puede explicar una parte de aquellos pacientes con la entidad a los cuales no se les detecta defecto genético, por lo que en este grupo particular de pacientes la búsqueda de dichos anticuerpos estaría indicada. La posibilidad de que existan otros autoanticuerpos contra otras proteínas del complemento se encuentra en investigación, así como otros genes y proteínas del complemento involucradas en su fisiopatología[5].
Se calcula que entre un 40 a 60% de pacientes con SHUa son portadores de mutaciones puntuales heterocigotas en genes del complemento (especialmente factor H y/o I), pero su penetrancia es aproximadamente del 50%, lo que explica que solo algunos portadores desarrollen la enfermedad. Ello, unido a que hasta un 10% de pacientes son portadores de más de un defecto y a que las variantes genéticas modulan la penetrancia, soporta la hipótesis de múltiples hits para el desarrollo de SHUa[19].
El diagnóstico de novo de SHUa en pacientes con trasplante renal ocurre en 1 a 5% de los casos, especialmente en los primeros tres meses luego del trasplante. Se considera que el uso de anticalcioneurínicos, ciertas infecciones virales, el rechazo vascular agudo donante cadavérico y la presencia de anticuerpos anti fosfolípidos positivos, son factores predisponentes en su aparición[7].
En aquellos casos en que la aparición de MAT ha sido asociada al uso de anticalcioneurínicos, se han utilizado varias estrategias de manejo (reducir dosis o cambiar de anticalcioneurínico) y la decisión final se debe tomar de manera individualizada. Lo que parece más claro es la mejoría de la MAT una vez se cambia o disminuye la dosis de inhibidor de calcineurina. Esto sugiere que, ante la persistencia de MAT cuando se suspende el medicamento, se debe considerar el diagnóstico de SHUa de novo en el pos trasplante, como en el caso del paciente tratado en esta publicación. La reposición de plasma puede utilizarse hasta mejorar el cuadro clínico, sin embargo, se ha visto que no cambia la historia natural de la enfermedad y el pronóstico en términos de morbimortalidad sigue siendo sombrío4. Así mismo, se ha visto que la infusión de plasma o el intercambio plasmático terapéutico puede producir una mejora hematológica transitoria, sin mayor impacto renal o en otros órganos comprometidos, como ocurrió en el paciente previamente expuesto. En pacientes con enfermedad renal crónica terminal que han sido trasplantados y han presentado SHUa, el factor de recurrencia de la enfermedad depende de la mutación que el paciente porta3, teniendo en cuenta que el hecho de no identificarla no excluye el diagnóstico ni el riesgo de recurrencia en el pos-trasplante[4].
En resumen, en el SHUa participan factores genéticos y ambientales y el defecto que ocasiona una regulación inadecuada del complemento se puede dar por pérdida de actividad (factor H, factor I), por mutaciones activadoras (factor B o proteína C3) o por autoanticuerpos (antifactor H), entre otras ya descritas. Este escenario induce a que, una vez activado el complemento (sea por gérmenes, fármacos, embarazo, células tumorales, etc.), éste no pueda ser regulado y el daño que ocasiona sobre las células endoteliales sea el directo responsable de la formación de trombos.
La sospecha diagnóstica de SHUa se inicia ante paciente con cuadro de MAT y, debido a que los signos y síntomas no permiten realizar el diagnóstico diferencial, es necesario realizar pruebas complementarias que permitan descartar otras MAT, permitiendo finalmente, por descarte, confirmar el diagnóstico clínico de SHUa. Para esto, se inicia con la solicitud de actividad de ADAMTS-13 con el propósito de descartar la presencia de PTT, y con el test de toxina Shiga en los casos de cuadro clínico compatible. Si este último es positivo, se está en presencia de un STEC-SHU y, si la actividad de ADAMTS-13 es < del 5%, ante una PTT, tal como se expuso previamente. Por último, si la actividad del ADAMTS-13 es > del 5% y la toxina Shiga es negativa, se está en presencia de un paciente con SHUa[3,4,9].
Es importante llegar a un diagnóstico preciso desde el punto de vista de la mutación, pues el pronóstico puede variar según el tipo de mutación, sin que este esté del todo claro en la literatura actual. Se ha visto cómo la alteración en el factor H tiene peor pronóstico, sin embargo, mutaciones del MCP que parecerían ser de mejor pronóstico, también tienen un alto impacto en la morbimortalidad a largo plazo, hecho posiblemente relacionado con la asociación con otras mutaciones. En el contexto de pacientes con enfermedad renal terminal, candidatos a trasplante, también es muy relevante establecer el perfil genético y evaluar el riesgo de recaída y la estrategia de manejo en el momento que ocurra el trasplante de órgano.
Aunque no se dispone de ensayos clínicos, la terapia plasmática (la infusión de plasma y el recambio plasmático) ha sido el tratamiento empírico de elección para pacientes con SHUa. Precisamente, nuestro paciente mejoró significativamente cuando recibió plasma para corregir alteraciones de coagulación antes de ser sometido a la biopsia renal[9,14]. Sin embargo, como se expuso previamente, el pronóstico global en términos de recurrencia, morbilidad y mortalidad no se ve impactado por este tipo de terapia.
La más reciente modalidad terapéutica para estos pacientes es el eculizumab. Se trata de un anticuerpo monoclonal humanizado que, al unirse con gran afinidad a la proteína C5 del complemento, impide su escisión en C5a y C5b. Con menor cantidad de C5a se disminuye la presencia de células inflamatorias y, más importante, con menor disponibilidad de C5b cae la generación del complejo terminal de ataque a la membrana (C5b-9)[15,20].
La eficacia y seguridad del producto ha sido evaluada, con la gran ventaja de que su respuesta es uniforme sin importar a que nivel esté la alteración, pues el fármaco actúa bloqueando C5, evitando así la generación de la C5-convertasa y por lo tanto la generación del complejo de ataque de membrana que es en parte responsable del daño endotelial y la generación de la MAT. Esto indica que el fármaco preserva las funciones del complemento en sus primeros pasos, que resultan cruciales en los procesos de opsonización de microorganismos y en la depuración de complejos inmunes circulantes y de cuerpos apoptóticos residuales. El uso temprano del eculizumab ha revelado no solo superioridad frente a la terapia con plasma, sino también mejor recuperación de la función renal, incluso si el paciente se encuentra en programa de hemodiálisis[16,20,22]. En nuestro paciente se evidenció mejoría marcada de la función renal y la calidad de vida con el inicio del tratamiento anti-complemento.
Debe recordarse que los pacientes con deficiencia constitucional de C5 presentan un riego incrementado de meningitis por Neisseria. Por ello, el uso de eculizumab se ha asociado al desarrollo de esta infección (alrededor de 4 casos por 1000 pacientes/año) y es la razón de la vacunación mandatoria de estos pacientes antes de la aplicación del fármaco[22,24].
Existe una relación entre la severidad de la etapa aguda y el pronóstico a largo plazo. Se asocian a peor pronóstico paciente con anuria mayor de 15 días, la presentación de manifestaciones extra renales, sobretodo neurológica, así como los pacientes con mutaciones en FH y C3. Por su parte, frente al antecedente de diarrea la demostración de la toxina Shiga mejora el pronóstico[17,25,26].
Conclusión
Se presenta el caso de un paciente con trasplante renal que ingresa por manifestaciones neurológicas. Luego de un prolongado y exhaustivo estudio, sus manifestaciones clínicas y hallazgos de laboratorio, compatibles con una MAT que no mejoro al suspender el inhibidor de calcineurina, habiendo descartado otras causas probables de MAT como la PTT y sin la presencia de rechazo en la biopsia renal, permitieron concluir que el paciente estaba desarrollando un SHUa de novo en el pos trasplante.
Si bien estas son patologías raras, la principal barrera médica es su desconocimiento y este caso permite ilustrar el ejercicio clínico para poder llegar a un diagnóstico definitivo. Todo paciente con la triada de anemia hemolítica, trombocitopenia e insuficiencia renal debe completar todo el ejercicio diagnostico exhaustivo de las MAT para poder llegar a un diagnóstico correcto y definir la terapéutica adecuada.
Responsabilidades éticas
Protección de personas y animales
Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos
Los autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informado
Los autores declaran que en este artículo no aparecen datos de pacientes.
REFERENCIAS
1. George JN, Charania RS. Evaluation of patients with microangiopathic hemolytic anemia and thrombocytopenia. Semin Thromb Hemost [Internet]. 2013 [accedido 01 Jan 2000];39(2):153-60. Available from: Available from: http://dx.doi.org/10.1055/s-0032-1333538 . Mayo 2016. [ Links ]
2. Polito MG, Kirsztajn GM. Thrombotic microangiopathies: thrombotic thrombocytopenic purpura / hemolytic uremic syndrome. J Bras Nefrol [Internet]. 2010 [accedido 01 Jan 2000];32(3):303-15. Available from: Available from: http://www.ncbi.nlm.nih.gov/pubmed/21103695\nhttp://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21103695 . Mayo 2016. [ Links ]
3. Campistol Plana JM, Arias M, Ariceta Iraola G, Blasco M, Espinosa M, Grinyo JM, et al. Actualización en síndrome hemolítico urémico atípico: diagnóstico y tratamiento. Nefrología (Madr.) [Internet]. 2013 [accedido 01 Jan 2000]; 33:27-45. Available from: Available from: http://dx.doi.org/10.3265/Nefrologia.pre2012.Nov.11781 . [ Links ]
4. Cordoba JP, Mariel K, Carolina C, Espitaleta Z, Gonzalez E, Ibarra M, et al. Síndrome hemolítico urémico atípico, revisión de la literatura y documento de consenso: enfoque diagnóstico y tratamiento. Rev Colomb Nefrol. 2015;2(1):19-40. [ Links ]
5. Kavanagh D, Goodship THJ. Atypical hemolytic uremic syndrome. Curr Opin Hematol. 2010; 17(5):432-8. [ Links ]
6. Zipfel PF, Wolf G, Jhon U, Kentouche K, and Skerka C. Novel developments in thrombotic microangiopathies: is there a common link between hemolytic uremic syndrome and thrombotic thrombocytic purpura? Pediatr Nephrol. 2011; 26: 1947-56. [ Links ]
7. Ali M, Syed A, Bhandari S. Case series: hemolytic uremic syndrome--another cause of transplant dysfunction. Transplant Proc [Internet]. 2013 [accedido 01 Jan 2000]; 45(9):3284-8. Available from: Available from: http://dx.doi.org/10.1016/j.transproceed.2013.07.060 . [ Links ]
8. Hirt-Minkowski P, Dickenmann M, Schifferli JA. Atypical Hemolytic Uremic Syndrome: Update on the Complement System and What Is New. Nephron Clin Pract [Internet]. 2010 [accedido 01 Jan 2000]; 114(4):c219-35. Available from: Available from: http://dx.doi.org/10.1159/000276545 . [ Links ]
9. Laurence J. Atypical Hemolytic Uremic Syndrome ( aHUS ): making the diagnosis. Clin Adv Hematol Oncol [Internet]. 2012 Oct [accedido 01 Jan 2000]; 10 (10 Suppl 17):1-12. Available from: Available from: https://www.ncbi.nlm.nih.gov/pubmed/23187605 . [ Links ]
10. Kreuter J, Winters JL. Drug-associated thrombotic microangiopathies. Semin Thromb He-most [Internet]. 2012 [accedido 01 Jan 2000]; 38(8):839-44. Available from: Available from: http://dx.doi.or-g/10.1055/s-0032-1328886 . [ Links ]
11. Vega J, Parodi C, Mendez GP, Goecke H. Síndrome hemolítico-urémico asociado al uso de gemcitabina. Rev Med Chil. 2013; 141(6):797-802. [ Links ]
12. Fakhouri F, Fremeaux-Bacchi V, Loirat C. Atypical hemolytic uremic syndrome: from the rediscovery of complement to targeted therapy. Eur J Intern Med [Internet]. 2013 [accedido 01 Jan 2000]; 24(6):492-5. Available from: Available from: http://dx.doi.org/10.1016Zj.ejim.2013.05.008 . [ Links ]
13. de Cordoba SR, Montes T. Síndrome hemolítico urémico atípico. Nefrol Supl Extraordin. 2011; 2(1):58-65. [ Links ]
14. Joseph C, Gattineni J. Complement disorders and hemolytic uremic syndrome. Curr Opin Pediatr [Internet]. 2013 Apr [accedido 01 Jan 2000]; 25(2):209-15. Available from: Available from: http://dx.doi.org/10.1097/MOP.0b013e32835df48a . [ Links ]
15. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med [Internet]. 2013 [accedido 01 Jan 2000]; 368(23):2169-81. Available from: Available from: http://dx.doi.org/10.1056/NEJMoa1208981 . [ Links ]
16. Rathbone J, Kaltenthaler E, Richards A, Tappenden P, Bessey A, Cantrell A. A systematic review of eculizumab for atypical haemolytic uraemic syndrome (aHUS). BMJ Open [Internet]. 2013 [accedido 01 Jan 2000]; 3(11): e003573. Available from: Available from: http://dx.doi.org/10.1136/bmjopen-2013-003573 . [ Links ]
17. Loirat C, Fremeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis [Internet]. 2011 [accedido 01 Jan 2000]; 6(1):60. Available from: Available from: http://dx.doi.org/10.1186/1750-1172-6-60 . [ Links ]
18. Kavanagh D, Goodship T. Genetics and complement in atypical HUS. Pediatr Nephrol [Internet]. 2010 [accedido 01 Jan 2000];25(12):2431-42. Available from: Available from: http://dx.doi.org/10.1007/s00467-010-1555-5 . [ Links ]
19. Kremer Hovinga JA, Vesely SK, Terrell DR, Lammle B, George JN. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood [Internet]. 2010 [accedido 01 Jan 2000];115(8):1500-11. Available from Available from http://dx.doi.org/10.1182/blood-2009-09-243790 . [ Links ]
20. Keating GM. Eculizumab: A Review of Its Use in Atypical Haemolytic Uraemic Syndrome. Drugs [Internet]. 2013 [accedido en 01 Jan 2000]; 73(18):2053-66. Available from: Available from: http://dx.doi.org/10.1007/s40265-013-0147-7 . [ Links ]
21. Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, Delmas Y, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int [Internet]. 2015 [accedido 01 Jan 2000]; 87(5):1061-1073 Available from: Available from: http://dx.doi.org/10.1038/ki.2014.423 . [ Links ]
22. Hillmen P, Muus P, Roth A, Elebute MO, Risitano AM, Schrezenmeier H, et al. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol [Internet]. 2013 [accedido 01 Jan 2000]; 162(1):62-73. Available from: Available from: http://dx.doi.org/10.1111/bjh.12347 . [ Links ]
23. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol [Internet]. 2007 [accedido 01 Jan 2000]; 25(11):1256-64. Available from: Available from: http://dx.doi.org/10.1038/nbt1344 . [ Links ]
24. Hillmen P. The role of complement inhibition in PNH. Hematology [Internet]. 2008 [accedido 01 Jan 2000]; 2008(1):116-23. Available from: Available from: https://doi.org/10.1182/asheducation-2008.L116 . [ Links ]
25. Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher M A, Niaudet P, Guest G, et al. Differential Impact of Complement Mutations on Clinical Characteristics in Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol [Internet]. 2007 [accedido 01 Jan 2000]; 18(8):2392-400. Available from: Available from: https://doi.org/10.1681/ASN.2006080811 . [ Links ]
26. Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, et al. Genetics of HUS : the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood [Internet]. 2006 [accedido 01 Jan 2000]; 108(4):1267-79. Available from: Available from: https://doi.org/10.1182/blood-2005-10-007252 . [ Links ]
Recibido: 07 de Noviembre de 2016; Aprobado: 10 de Enero de 2016; Publicado: 23 de Febrero de 2017
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
