











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Systemic vasculitis associated with anti-neutrophil cytoplasmic antibodies (ANCA) include Churg-Strauss syndrome, Wegener's granulomatosis (WG), microscopic polyangiitis (MPA), and its limited renal form: idiopathic necrotizing glomerulonephritis[1]. WG is characterized by necrotizing granulomatous small-vessel vasculitis that usually affects the upper respiratory tract, lungs and kidneys.
The disease was first described in the medical literature at the end of the 19th century in a clinical case. Then, in the 1930s, it was named by Friedrich Wegener, who described the clinical triad consisting of lung, kidney and upper respiratory tract compromise. Since 2011, WG is known as granulomatosis with polyangiitis (GPA)[2]. The disease may occur in a localized manner, generally affecting the upper respiratory tract, or systemically, which is more severe and aggressive. Clinical manifestations depend on the involvement of compromised blood vessels and, in addition to the classic triad, may also affect the central and peripheral nervous system, skin, gastrointestinal tract and musculoskeletal system.
Incidence of the disease is between 2 and 12 cases per million inhabitants, equally frequent in men and women. It is very rare in children and young adults. Peak incidence is reported in the seventh decade of life, between 65 and 70 years of age, with a prevalence of 24-157 cases per million inhabitants[2,3]. Mortality incidence rate is 43.5 (95% CI: 35.4 to 52.9) per 1000 people/year. In the first year after diagnosis, the rate is 97.4 (96% CI: 69.6 to 132.6) per 1000 people/year; however, as the years pass, mortality decreases[4]. Patients with renal impairment have an increased risk of mortality, especially those requiring dialysis therapy, since mortality increases with the passage of time. Its main causes are cardiovascular events and infections[5,6].
The case of a patient with WG treated at the Hospital San José (Bogotá, Colombia), and a review of the literature focused on the renal involvement of the pathology are presented below.
Case report
A 76-year-old female patient with a history of chronic hypertension, hypothyroidism, chronic venous insufficiency and glaucoma consulted the Hospital San José emergency room for a 1-month clinical picture of intermittent fetid bilateral otorrhea -associated with rhinorrhea-, cough with whitish expectoration, hyporexia and fever. The patient was examined by the otorhinolaryngology service; non-fetid detritus was observed in the auditory ducts during the bilateral otoscopy. In the anterior rhinoscopy, nasal crusts were observed, so she was diagnosed with active chronic otitis media and acute rhinosinusitis. Consequently, physicians ordered her hospitalization and initiated management with intravenous ampicillin-sulbactam and dexamethasone-ci-profloxacin (Fixamicin® Dexacipro) in ear drops.
The patient presented respiratory distress and tachycardia, while laboratory tests reported leu-kocytosis, anemia, elevated nitrogen compounds (creatinine: 2.7 mg/dl, BUN: 36 mg/dl), and chest x-ray with alveolar opacities involving the perihilar and basal regions of both lungs by multilobar consolidation and free bilateral pleural effusions in small quantities. Internal medicine assessment was requested; she was examined and diagnosed with severe community-acquired pneumonia, with CURB-654, multilobar involvement and AKIN II. Thus staggered antimicrobial therapy of piperacillin/tazobactam and vancomycin was administered; also, oseltamivir is added to the management after suspected viral pneumonia due to H1N1.
On the second day of hospitalization, the patient presented deterioration of the clinical picture, with type 1 acute respiratory failure. Orotracheal intubation, onset of mechanical ventilation, vasopressor support and transfer to an intensive care unit were performed. Chest CT was requested; areas of increased lung parenchyma density with a ground glass pattern and bilateral multilobar consolidations of greater profusion in the upper lobes (especially in the upper left lobe) and free bilateral pleural fluid were found. The pulmonology service was ordered a fibro-bronchoscopy with bronchoalveolar lavage report: macrophages: 67%; lymphocytes: 0%; polymorphonuclear leukocytes: 23%; Giemsa: negative for Pneumocystis jirovecii; Ziehl-Neelsen stain: negative for AARB; Grocott: structures with Candida yeast and pseudohypha morphology. B smears show presence of ciliated cells, alveolar macrophages and occasional polymorphonuclear leukocytes on a clean background. Furthermore, since there were no tumor cells in this material, the pneumology service decided to continue antibiotic regimen and added antifungal management with fluconazole to the treatment.
The medical service observed deterioration of renal function (creatinine: 2.9 mg/dl, BUN: 51 mg/ dl); interconsultation with nephrology is requested after suspicion of acute tubular necrosis. It is believed that, in the context of a patient with a history of upper respiratory tract infection, anemia and impaired renal function, small vessel vasculitis should be considered as the first possibility (Wegener's granulomatosis vs microscopic polyangiitis). Therefore, urinalysis was requested, reporting: density: 1015; pH: 5, negative nitrites; proteins: 150; normal glucose; epithelial cells: 0-2xc; scarce bacteria; leukocytes: 0-2xc; red cells: 20-30xc; mucus +; and crystals of amorphous urates ++. No bacteria were observed in the urine Gram. 24-hour urine protein test reported 0.7 gr/24h. Renal and urinary tract ultrasonography showed kidneys of usual location and size, with adequate cortical thickness and good corticomedullary differentiation, with no evidence of intraparenchymal focal lesions. There was a diffuse bilateral increase in renal parenchyma echogenicity.
An immunological test was also ordered, reporting positive results for c-ANCA antibodies, with titer 1:160 and direct Coombs 2+. Complement C3, Complement C4, anti-La/SSB, anti-RNP, anti-Ro/ SSA, anti-SLC-70 ac, anti-Sm (anti Smith), creatine-kinase MB (CK-MB) and p-ANCA (perinuclear anti-neutrophil cytoplasmic antibodies) tests were within the normal range.
With the positive c-ANCA report, in addition to the signs and symptoms informed by the patient and rapid deterioration of respiratory, renal and hemodynamic conditions, the diagnosis of WG type small vessel vasculitis was confirmed, thus boluses of methylprednisolone for 3 days were administered on the 14th day of hospitalization. A renal biopsy was also ordered with immunofluorescence tests to characterize renal involvement due to proteinuria and diffuse parenchymal renal disease.
Hematology service was interconsulted for suspicion of hemolytic anemia by positive direct Coombs test and peripheral blood smear report, with normochromic normocytic red cells, polychromatophilia: 1+, schistocytes: 1+, anisocytosis: 3+. The specialty considered that normochromic normocytic anemia was not of autoimmune origin, but given the aggressive progression of the clinical picture, hematolymphoid neoplasia processes had to be ruled out; therefore, bone marrow and protein electrophoresis tests were requested, reporting absence of monoclonal peak in protein electrophoresis. With these results, the hematology service ruled out neoplastic pathology as the cause of anemia.
The renal biopsy report was received; renal parenchyma with seven glomeruli: four of them with global sclerosis and three viable with extracapillary proliferation in the fibrocellular phase, endocapillary proliferation and leukocyte entrapment; there was no necrosis or karyorrhexis. Tubules showed 20% atrophy, associated with fibrosis in the same percentage, with diffuse mononuclear inflammatory cell infiltrate. There was no thrombosis, vasculitis or granulomas in the vessels. Immunofluorescence of 14 glomeruli, 2 baseline IgG sclerosed glomeruli, C1q, negative c3, focal accumulations in IgM sclerosed glomeruli, negative IgA, linear kappa in a glomerulus, linear lambda in a glomerulus, negative albumin. Findings showed pauci-immune extracapillary proliferative glomerulonephritis with changes in chronicity.
Upon completion of pulse steroid therapy, management was continued with prednisolone (dose of 30 mg/day orally) and cytostatic management with rituximab (dose of 375 mg/mt2/week) for 4 weeks was added. Two days after initiation of oral prednisolone therapy, the patient presented altered state of consciousness associated with elevated nitrogen compounds, so it was considered that she had uremic encephalopathy (BUN: 118 mg/dl, creatinine: 3.4 mg/dl). Hence initiation of renal replacement therapy was indicated in the form of conventional hemodialysis, with a duration of 3 to 4 hours per day. The procedure was performed daily, improving the state of consciousness and signs of water intoxication. The patient was extubated on the fourth day of renal replacement therapy; her evolution was satisfactory and she was transferred from ICU to the general ward, where dialysis every other day, immunosuppressive therapy with prednisolone, and scheduled doses of rituximab were continued.
The pathology test confirmed the diagnosis: Wegener's Granulomatosis-type small vessel vasculitis. She was discharged upon stabilization of clinical picture and continued outpatient renal replacement therapy plus prednisolone (dose of 30 mg/day orally).
Literature review and discussion
Etiopathogenesis
WG usually begins as a granulomatous disease of the airways that progresses to systemic vasculitis, suggesting an aberrant cell-mediated immune response to (exogenous or endogenous) antigens and resulting in the formation of granulomas.
Autoimmune etiopathogenesis is complex, since it involves the production of ANCA directed to proteinase 3 (PR3) in approximately 80% of patients and directed to myeloperoxidase (MPO) in approximately 10%. Lysosome associated membrane protein 2 (LAMP-2) antibodies may also play a role in pathogenesis through a molecular mimicry process. In vitro and animal models, using different approaches, support the idea that an interaction of PR3-AN-CA with PR3 released from azurophil granules and expressed on the cell surface of TNF-a-activated neutrophils results in the premature degranulation of neutrophils, with subsequent endothelial damage and leukocyte recruitment[2].
On the other hand, infections (especially Staphylococcus aureus), the environment, and some chemicals, toxins or drugs have been suggested as triggers of the disease in people with genetic predisposition[7].
Clinical manifestations
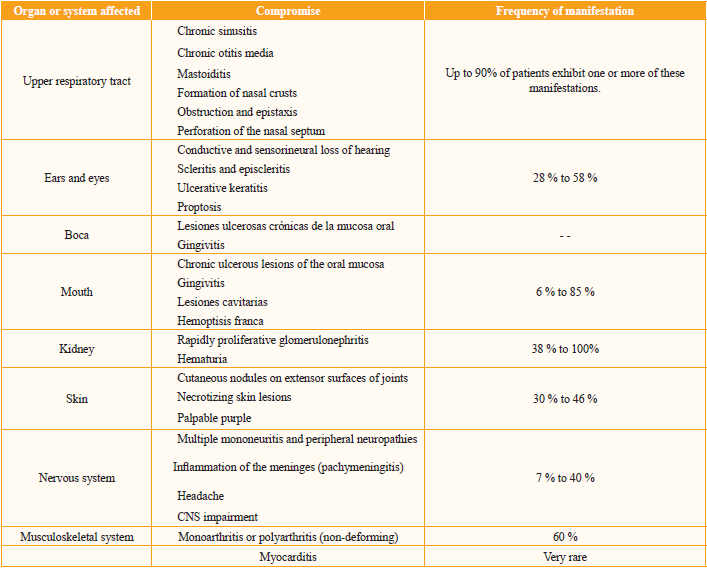
The manifestation spectrum tends to be very heterogeneous, more commonly in ears, nose and throat[8]. Clinical manifestations often vary with the stage of disease and the degree of involvement of the organ[9]. Table 1 shows compromised systems, as well as involvement and frequency of manifestation.
Kidney disease is sometimes the initial manifestation, or occurs in the course of disease. It may progress from asymptomatic and mild forms to fulminant glomerulonephritis in a matter of days or weeks, resulting in end-stage renal failure; even with appropriate therapy, the disease may lead to chronic renal failure[10]. Hence, up to 30% of patients with moderate to severe renal disease at the time of diagnosis will require renal replacement therapy. Between 40% and 70% of patients may recover renal function following induction therapy[11].
To measure disease activity, the use of tools such as the Birmingham Vasculitis Activity Score for Granulomatosis with Polyangiitis (BVAS/GPA), which categorizes organ involvement and disease activity in each system, has been proposed, defining activity as abnormalities caused by the newly diagnosed disease or worsening in the last four weeks. It evaluates disease activity at a given point in time as the sum of the manifestations of the individual organ system, defined by a list of 34 weighted elements caused by the active disease with respect to whether the symptoms are new, worse, or persistent. For evaluation, each of the 34 items is classified as either major or minor, depending on immediate threat to life or organ, with a score of 3 to 1 and a combined maximum score of 68. The ascent values of this score are a negative predictor of survival.
When assessing disease activity, consideration should be given to what is inherent to the active disease and to the permanent damage caused by the disease. Abnormalities produced by vasculitis that persist for more than four weeks are termed persistent disease and give rise to damage that is defined as irreversible changes resulting from scarring and present for at least three months.
Diagnosis
Diagnosis is based on suggestive clinical symptoms. The gold standard is the histological test where necrotizing vasculitis of small vessels and granulomatous inflammation with multinucleated giant cells are observed.
Histopathological identification is seldom performed in the early stages of WG. In patients with clinical symptoms for which biopsy is not possible or not diagnostic, positive ANCA results help in the diagnosis of these cases. ANCAs are antibodies whose targets are the two main components of neutrophil granulocytes: serine PR3 and myeloperoxidase. Anti-serine PR3 antibodies are practically pathognomonic of WG, whereas anti-myelo-peroxidase antibodies are more suggestive of other primary necrotizing vasculitis, mainly microscopic polyangiitis[2].
There are two types of tests to detect ANCA: immunofluorescence and enzyme-linked immunosorbent assay (ELISA). Immunofluorescence distinguishes between anti-PR3 and anti-myeloperoxidase based on its staining pattern: the former is associated with c-ANCA and the second with p-ANCA. Estimation of ANCA titers in serum using immunofluorescence techniques provides reliable diagnosis, often with no need for a positive biopsy. The sensitivity of c-ANCA titers depends on the disease activity2. Twenty percent of patients with active WG have negative ANCA. This percentage increases to 30% in the localized forms of the disease[2].
Treatment
Current therapies minimize local and systemic inflammation and can preserve organ function. Immunosuppressive agents are combined with support management, including: compensation for organ dysfunction (treatment of hypertension or dialysis); treatment or prevention of comorbidity (infection, osteoporosis, or cataracts); worsening of preexisting comorbidity (worsening of ischemic heart disease or obesity), or development of new pathologies. Approach to the patient should include assessment of the severity and context in which the disease occurs. Table 2 shows the immunosuppressive treatments used.
Table 2 Drugs frequently used in the treatment of Wegener's Granulomatosis
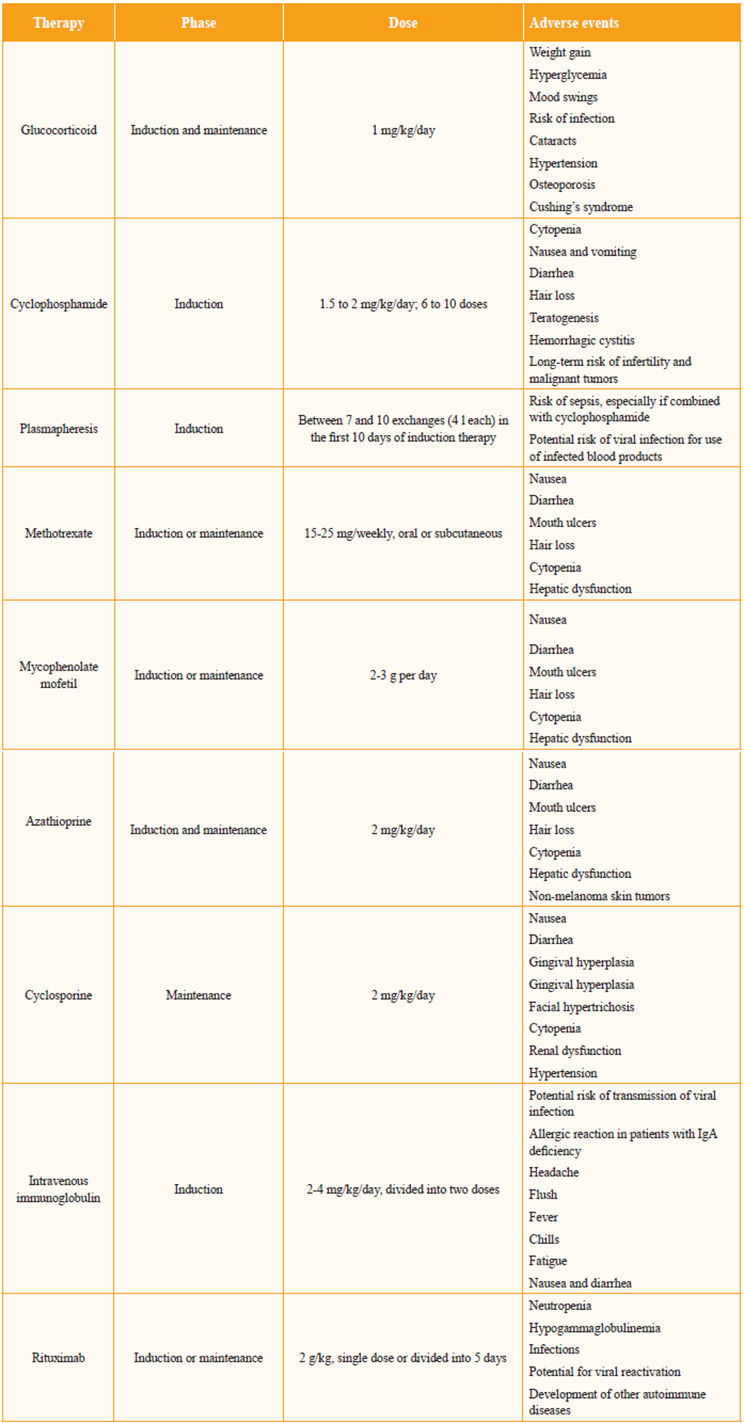
Source: Modified from Tarabishy AB, Schulte M, Papaliodis GN, Hoffman GS. Surv Ophthalmol. 2010; 55:429-4422.
Generally, patients with WG respond well to immunosuppressive therapy with cyclophosphamide or methotrexate, associated with glucocorticoids[12]. The current recommended dose for cyclophospha-mide is 1.5 to 2 mg/kg/day and for prednisone 1 mg/ kg/day orally[13].
Patients receive intensive induction therapy followed by maintenance therapy. Most patients will require additional therapy to control comorbidity and limit drug toxicity. Induction treatments may be repeated for relapse; however, it may be necessary to change the type of induction due to toxicity or poor initial response.
During the remission stage of the disease, over the first month, prednisone is titrated up to 5 mg/ week. This regimen should be followed for one year after complete remission, and then gradually reduced until discontinuation. Complete remission can take from a few months to 1 or 2 years, with an average time of 12 months. Using the cyclophosphamide and corticosteroid regimen, significant improvement was observed in more than 90% of the patients, achieving remission of the disease in 75% of the cases. However, relapses have been observed in up to 50% of patients in remission13.
Methotrexate and azathioprine have been used in the recovery phase with good response; however, methotrexate should not be used in patients with a glomerular filtration rate of 50 ml/min or less. Glucocorticoid boluses have also been frequently used to control reactivations. Mycophenolate, deoxys-pergualine and leflunomide are other drugs that have been shown to be promising in the maintenance of remission.
Rituximab has recently been used during induction and remission. In 2014 the National Institute for Health and Care Excellence (NICE) recommended the use of rituximab with a glucocorticoid in the following cases: if treatment with cyclophosphamide exceeds the maximum cumulative dose; if contraindicated or not well tolerated; if a possible alteration of fertility is anticipated; if the disease has been active or progressed despite a course of 3 to 6 months of treatment; if the person had urothelial neoplasia14. These recommendations were taken from two studies: RAVE and RITUXVAS15, 16. The RITUXVAS study enrolled patients with renal involvement, who were randomly assigned to rituximab plus cyclophosphamide (n = 33) or cyclophosphamide (n = 11). Both groups received methylprednisolone intravenously (1 g) and an oral glucocorticoid (1 mg/kg/day initially, reduced to 5 mg/day after 6 months).
Patients in the rituximab group received infusions of 375 mg/m2 per week for 4 weeks and 15 mg/kg of intravenous cyclophosphamide with the first and third infusions of rituximab. Patients in the rituximab group received no maintenance treatment. Patients in the control group received intravenous cyclophosphamide (15 mg/kg for 3 to 6 months, 6 to 10 doses in total), followed by azathioprine (2 mg/kg/day) as maintenance. Patients were also allowed treatment with rituximab or cyclophosphamide if they relapsed.
Sustained remission occurred in 76% of patients in the rituximab plus cyclophosphamide group and in 82% of patients in the cyclophosphamide group. The absolute difference in sustained remission with rituximab plus cyclophosphamide, compared with cyclophosphamide, was -6% (95% CI: -33 to 21). Among patients who were still in the study after 12 months, 93% of those in the rituximab plus cyclophosphamide group and 90% of patients in the cyclophosphamide group were in sustained remission. No benefit of rituximab on other immunomodulators was found.
Another randomized clinical trial compared treatment during the remission phase of rituximab (n = 57) and azathioprine (n = 58) after receiving cyclophosphamide and glucocorticoid management. The groups received 500 mg of rituximab on days 0 and 14 and 6, 12 and 18 months after enrollment in the study or azathioprine every day until month 22. The authors demonstrated the superiority of rituximab (29% versus 3% p = 0.000), with similar frequency of adverse events in both groups (n = 25) (17).
The Clinical Practice Guideline for ANCA-associated vasculitis with renal impairment makes recommendations for treatment of severe systemic GW18, based on combined treatment: methylprednisolone and cyclophosphamide pulse therapy as first-line treatment to achieve remission. It does not recommend the use of long-term cyclophosphamide, given the increased risk of relapse and notes that consideration should be given to pre- and post-infusion white blood cell counts. Recommendation for the use of rituximab is the same as that contained in the NICE guidelines. The use of methotrexate and glucocorticoid may be given as an alternative to cyclophosphamide in early systemic diagnosis and GFR > 60 ml/min/1.73m2 of body surface area, but is less effective in disease control (relapse) during maintenance. In renal and/or hepatic failure, the use of methotrexate is contraindicated, in which case mycophenolate plus glucocorticoid is an alternative therapeutic option. Plasmapheresis, as an adjuvant therapy, would be indicated in severe cases with rapidly proliferative glomerulonephritis to improve renal survival or dialytic requirement and/or pulmonary hemorrhage.
Intravenous immunoglobulins (IVIg) have demonstrated potent therapeutic efficacy in patients with positive ANCA vasculitis; they are considered as a therapeutic alternative in patients with refractory disease or in patients for whom conventional therapy is contraindicated, e.g. when there is infection, in severely ill patients, or during pregnancy. As of now, there are no guides on duration, frequency or optimal dose of IVIg; some authors use this regimen based on small published series, associating it with corticosteroids, until immunosuppressive agents are no longer contraindicated8.
A Cochrane review identified a randomized clinical trial (RCT) involving 34 participants who were assigned to receive IVIg (a single cycle of 400 mg/kg/day for 5 days) or placebo, in addition to azathioprine and systemic corticosteroid for remission maintenance19. It was found that there were no significant differences when comparing adjuvant IVIg with adjuvant placebo in mortality outcomes, adverse events, time to relapse and infection. The decrease in disease severity derived from patient-reported symptoms was slightly higher in the IVIg group than in the placebo group, after one and three months. There was also a significant increase in total adverse events in the IVIg group (relative risk: 3.50 95% CI: 1.44 to 8.48; P <0.01). The authors concluded that the RCT does not provide sufficient evidence to assure that adjuvant therapy with IVIg provides a therapeutic advantage compared to the combination of steroids and immunosuppressants for patients with WG.
Plasmapheresis has been recognized as a second-line treatment. According to the American Society of Apheresis (ASFA), plasmapheresis is a class I indication for WG in dialysis dependent cases and category III in non-dialysis-dependent cases20. Malhotra et al.21 reported the case of a non-dialysis dependent patient who underwent joint treatment of plasmapheresis with rituximab and achieved remission after failure to respond to high-dose cyclophosphamide therapy. For this patient, when there was no improvement with cyclophosphamide, it was decided to start rituximab. However, since the peak of action of rituximab occurs at 3-4 weeks after administration, plasmapheresis was started for acute and critical period management.
The erythrocyte sedimentation rate (ESR) and c-ANCA levels are used to monitor disease activity and early diagnosis of relapse; however, there is some discussion about its true usefulness.
WG can be associated with early mortality, especially in patients with renal impairment who do not receive early immunomodulatory treatment. Several treatment modalities have been employed depending on the compromised organs and systems. Although the use of cyclophosphamide plus systemic steroid therapy is recommended as initial therapy, adverse events and high relapse rate do not appear to be the best therapy for systemic use; moreover, doses and duration of doses are not well defined.
In our patient, a classic manifestation of chronic airway compromise, bilateral lung involvement and progressive renal failure were observed. The clinical and paraclinical findings showed an infectious systemic compromise, which could aggravate the renal compromise. The empirical antibiotic staggering was adequate, according to institutional recommendations; however, the suspicion of autoimmune disease stated by the nephrology service has a significant impact on the evolution of the patient. Although the use of cyclophosphamide is considered the first-line treatment, it is important to assess the context of each patient (individualizing). In our case, we opted for the use of rituximab to avoid compromising cellular immunity, since the patient manifested neutropenia.
The optimal maintenance of remission in granulomatosis with polyangiitis (GPA) and other ANCA-associated vasculitis (AAV) is still under debate. Due to renal compromise, we consider that rituximab treatment in our patient is an excellent choice that is in accordance with the literature review, representing a lower risk of relapse and secondary complications.
The treatment of vasculitis has improved, but until the disease can be completely controlled or cured, it remains unsatisfactory. It is possible to prevent early mortality and the immediate effects of active vasculitis on the function of the organ have been reduced. However, our goal is to further increase the likelihood of survival, as well as to improve the quality of life of patients who survive severe manifestations, ensuring minimization of disease activity and damage, drug toxicity and deterioration of the quality of life.
Conclusions
Renal impairment in patients with WG is common and may become lethal. A high proportion of patients may require renal replacement therapy, although many of them are able to return to normal renal function. The factors that are associated with the return of normal renal function are unclear but have been reported to be dependent on early management and high doses of immunomodulators.
Now, in relation to treatment regimens, they have been based on clinical trials with a small number of participants and on observational studies with risk of bias, so the ideal treatment (lower rate of relapse, lower adverse effects, lower mortality, greater healing) is not yet fully defined. Rituximab is a drug that is being used on a larger scale than other conventional treatments, with adequate effectiveness and minor side effects in these patients. It should be noted that two clinical practice guides have clear recommendations about its uses, although such recommendations do not apply to all patients, especially in the case managed by our institution. Therefore, we consider that more studies are needed to evaluate rituximab treatment in patients with severe compromise and other comorbidities

