Los haplotipos colombianos de la mutación N370S causante de la enfermedad de Gaucher pueden provenir de un haplotipo ancestral común
Ricardo Wilches 1, Hugo Vega 2, Olga Echeverri 1, Luis Alejandro Barrera 1
1 Instituto de Errores Innatos del Metabolismo, Facultad de Ciencias, Pontifica Universidad Javeriana, Bogotá D.C., Colombia.
2
Instituto de Genética, Universidad Nacional de Colombia, Bogotá D.C., Colombia.Introducción. La enfermedad de Gaucher es una condición panétnica, caracterizada por la acumulación de glucosilceramida en los macrófagos. La causa principal de esta enfermedad en algunos países occidentales, incluido Colombia, es la mutación N370S, en el gen de la glucocerebrosidasa localizado en 1q21.
Objetivo. Determinar el grado de asociación entre la mutación N370S y los alelos de cinco microsatélites cercanos al sitio de la mutación en nueve pacientes colombianos Gaucher tipo 1, procedentes del altiplano cundiboyacense.
Materiales y métodos. A partir del ADN de los pacientes, sus familiares cercanos y 30 individuos control, los loci: D1S305, D1S2624, D1S2777, ITG6.6.2 y 5GC3.2 fueron amplificados mediante reacción en cadena de la polimerasa. Las frecuencias alélicas por microsatélite fueron calculadas en pacientes y controles y 11 haplotipos N370S fueron inferidos y se determinó el grado de desequilibrio de ligamiento entre los alelos de cada haplotipo con la mutación N370S.
Resultados. Se encontró un haplotipo consenso N370S compuesto por los alelos 222-314-260-301-172 (pares de bases) que corresponden a los microsatélites: 5GC3.2 ITG6.6.2, D1S2777 D1S2624 y D1S305 respectivamente. Hubo desequilibrio de ligamiento significativo entre los alelos de 222, 314, 260 y 301 pares de bases y la mutación N370S.
Conclusión. Una fracción conservada del haplotipo pudo haber estado asociada la mutación en un cromosoma ancestral al grupo de pacientes, cuya procedencia étnica es aún desconocida.
]]> Palabras clave: enfermedad de Gaucher, mutación, haplotipos, ligamiento (Genética), reacción en cadena de la polimerasa.Colombian haplotypes of the Gaucher disease-causing N370S mutation may originate from a possible common ancestral haplotype
Introduction. Gaucher disease is a pan-ethnic condition characterised by glucosylceramide accumulation in macrophages due to glucocerebrosidase deficiency. Its gene, GBA, has been mapped to 1q21 and mutation N370S is the main cause of the disease in western populations, including Colombia.
Objective. To asses the degree of association between N370S mutation and the alleles of five microsatellites near the mutation site in the GBA locus in nine Colombian Gaucher patients, from the Cundinamarca-Boyacá region.
Materials and methods. DNA from patients bearing the N370S mutation, their closest relatives, and 30 controls was taken to PCR-amplify the markers: D1S305, D1S2624, D1S2777, ITG6.6.2 and 5GC3.2. Allele frequencies were calculated, haplotypes inferred and linkage disequilibrium levels between marker alleles and N370S were also estimated
Results. Eleven N370S chromosomes were obtained. A consensus N370S haplotype consisting of the alleles: 222-314-260-301-172 (base pairs) was identified. Each allele corresponding to markers 5GC3.2, ITG6.6.2, D1S277, D1S2624 and D1S305, respectively. There was statistically significant linkage disequilibrium between the alleles of 222, 314, 260, 301 base pairs and the N370S mutation.
Conclusion. A conserved fraction of the haplotypes suggests that N370S may be present among patients and stem from a single ancestral chromosome for which the ethnic origin is still unclear.
Key words: Gaucher disease, mutation, haplotypes, linkage (Genética), polymerase chain reaction.
La ausencia o reducción drástica de la actividad de la hidrolasa lisosomal b glucosidasa ácida (E.C. 3.2.1.45) constituye la principal causa bioquímica de la condición autosómica recesiva denominada enfermedad de Gaucher (EG). La variante menos grave de esta enfermedad (EG tipo 1) se caracteriza por hepatoesplenomegalia y lesiones esqueléticas, mientras las variantes tipo 2 y 3 aguda y sub-aguda, respectivamente, presentan además compromiso del sistema nervioso central (1).
Se han reportado más de un centenar de mutaciones en el gen de la glucocerebrosidasa (GBA); sin embargo, pocas mutaciones se encuentran con alta frecuencia en poblaciones como la judía asquenazí, las europeas no judías y otros grupos con ancestros europeos (1,2).
]]> Las mutaciones N370S (1226A®G) y 84GG (84-85insG) representan más del 90% de los alelos causantes de la enfermedad en judíos asquenazí (3,4), en tanto que las N370S y L444P (1448T®C) se encuentran comúnmente en europeos no judíos (5). La frecuencia de N370S en los distintos grupos de pacientes fluctúa entre 22 y 54%, seguida por L444P (23%).En Colombia y Argentina más del 40% de las mutaciones identificadas en los pacientes corresponde a N370S (6,7). La composición étnica de los pacientes argentinos incluye algunos con ancestros judíos asquenazí, mientras que los pacientes colombianos, salvo pocas excepciones, no refieren tener ancestros judíos o europeos. Las demás mutaciones halladas en pacientes argentinos (6) y colombianos son L444P y RecNciI, con frecuencias variables en ambos grupos de pacientes. En Colombia se han encontrado tres mutaciones nuevas (7).
El gen GBA, en la región 1q21, está localizado entre los marcadores moleculares tipo microsatélite D1S305 y D1S2624, los cuales enmarcan un fragmento de aproximadamente 2,2 mega bases (Mb) (8). Dentro de este fragmento, y ligados al gen GBA, están los marcadores D1S2777, D1S2721, ITG6.2, 5GC3.2 D1S2140 más el gen de la enzima piruvato cinasa (PKLR) (9,10).
Estos marcadores se han empleado en la construcción y análisis de haplotipos para las mutaciones N370S y 84GG en judíos asquenazí y europeos no judíos (10-12).
Con base en las frecuencias y la magnitud de las asociaciones de los alelos de los marcadores que constituyen los haplotipos, en estos estudios se ha sugerido que la mutación N370S apareció en un cromosoma ancestral común a todos los que actualmente la portan en las comunidades europeas judías y no judías. El fundamento teórico de esta conclusión reside en el concepto de desequilibrio de ligamiento, que se define como una medida o estimativo de la asociación entre los alelos de loci que están ubicados en el mismo cromosoma. Gracias a esta herramienta se han estudiado los alelos que constituyen un haplotipo. En el caso específico de los haplotipos N370S, se ha propuesto que la mutación se originó en un ancestro europeo no judío y que para el momento de la fundación del asquenazismo (alrededor del año 900 d.C.), un cromosoma portador de la mutación ingresó al acervo genético del naciente grupo judío. Debido a fenómenos de deriva genética y de la endogamia propia de este grupo, el haplotipo portador de la mutación N370S incrementó su frecuencia en esta población (10). A partir de la información obtenida durante los estudios moleculares iniciales con pacientes colombianos afectados por la enfermedad de Gaucher se observó que la mayoría de aquellos que portan la mutación N370S procede de una misma región del país: el altiplano cundi-boyacense (7). Dicho dato motivó el presente estudio, en el cual se pretende identificar los alelos de microsatélites ligados al gen GBA en asociación con la mutación N370S y establecer los respectivos haplotipos en el grupo de pacientes. Dependiendo de la constitución alélica de los haplotipos que se infieran será posible determinar si existe un haplotipo consenso que pueda reflejar el haplotipo N370S ancestral hipotético, el cual pudo haber aparecido en la zona de procedencia de los pacientes con el arribo de sus primeros habitantes, antes o después del periodo de dominación española (entre los siglos XVI y XIX).
Como un primer paso en la prueba de esta hipótesis se emplearon los microsatélites de la región 1q21: 5GC3.2, ITG6.2, D1S2777, D1S2624 y D1S305 para inferir los haplotipos N370S y determinar el grado de desequilibrio de ligamiento entre la mutación y los alelos identificados por marcador en nueve pacientes colombianos con enfermedad de Gaucher (EG) cuyas familias provienen del altiplano cundiboyacense.
Materiales y métodos
Previo consentimiento informado y aprobación por parte del Comité de Investigación y Ética de la Facultad de Ciencias de la Universidad Javeriana, se tomaron muestras de sangre total de nueve pacientes colombianos con enfermedad de Gaucher no relacionados entre sí y de familias provenientes del altiplano cundiboyacense, cuyos genotipos se establecieron previamente y que presentaron la mutación N370S en estado homoalélico o heteroalélico. También se tomaron muestras de sus familiares más cercanos y de 30 individuos normales que sirvieron de control para el análisis de desequilibrio de ligamiento. Las muestras de los familiares cercanos se tomaron con el fin de establecer la fase gamética que permitió inferir los haplotipos. El ADN se aisló usando el DNA purification kit (Promega, Madison, WI, USA). Los microsatélites D1S305, D1S2777, D1S2624, 5GC3.2 e ITG6.2 se amplificaron para cada individuo. Las condiciones de reacción en cadena de la polimerasa (PCR) se unificaron para todos los microsatélites de acuerdo con Cormand et al. (11) y Lau et al. (13).
Gracias a los fluorocromos con que fueron marcados los iniciadores forward de cada marcador, los alelos se identificaron en un secuenciador automático AB Prism 310 (PE Applied Biosystems Foster City, CA, USA). La información alélica, o sea los tamaños de los fragmentos amplificados en pares de bases, se procesó con el programa Genescan 3.1.2 y los genotipos por marcador para cada individuo se determinaron mediante el programa Genotyper 2.5.
Luego se determinaron las frecuencias alélicas tanto en pacientes como en controles y se estableció la fase gamética con la mutación N370S. Se siguió la nomenclatura alélica de CEPH - Généthon (14). Aunque la mayoría de los alelos de los microsatélites tiene una nomenclatura propia, se identificaron todos los alelos (los cuales se nombran en este artículo) en pares de bases para evitar confusiones con los que no la tienen.
]]> El análisis de desequilibrio de ligamiento se llevó a cabo por medio del parámetro d, que está diseñado para contrastar la frecuencia de un determinado alelo de un locus polimórfico en cromosomas portadores de una característica genética determinada,(sea esta una mutación o un alelo patogénico), con la del mismo alelo en cromosomas que expresan la forma silvestre de la condición (15,16).
En la ecuación anterior, PD es la frecuencia del alelo estudiado en los cromosomas portadores del alelo causante de la enfermedad y PN es la frecuencia del mismo alelo en el grupo de cromosomas control. Un valor d = 1 implica el máximo DL, mientras que un valor de 0 implica la no existencia de DL, valores de d < 0 se asumen como 0. La significación estadística de la asociación alélica se obtuvo por medio una prueba de ji cuadrado con un grado de libertad a partir del uso de tablas 2 x 2 (15).
Resultados
Haplotipos
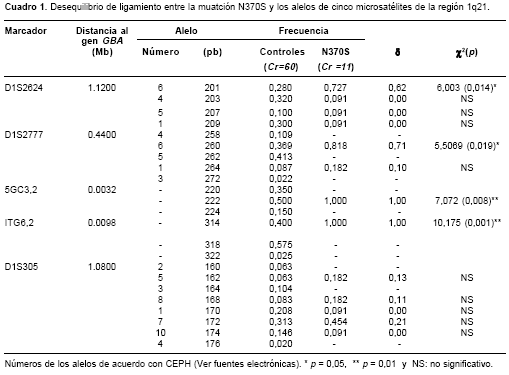
Se determinaron los alelos de los microsatélites en fase con la mutación N370S en los nueve pacientes. A partir de estos cromosomas con fase se establecieron las frecuencias alélicas por marcador, las cuales se muestran junto con las de los controles en el
cuadro 1.
De los nueve pacientes, siete fueron N370S heteroalélicos y los dos restantes N370S homoalélicos, estos dos últimos hijos de padres no consanguíneos. En total se infirieron once haplotipos N370S (
cuadro 2) ]]>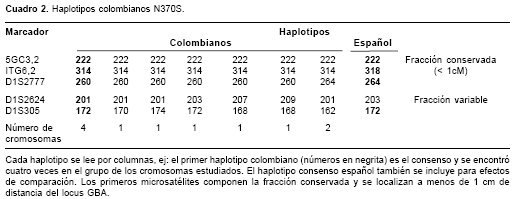
De acuerdo con las distancias entre cada microsatélite y el sitio del gen GBA en estos haplotipos se establecieron dos fracciones, una denominada "conservada" y la otra "variable". Los microsatélites 5GC3.2, ITG6.2 y D1S2777 componen la fracción conservada, mientras que D1S305 y D1S2624 constituyen la variable.
Con respecto a la fracción conservada, los once haplotipos están compuestos por los alelos de 222 y 314 pares de bases (pb) correspondientes a los microsatélites 5GC3.2 e ITG6.2. Cuando se tiene en cuenta el marcador D1S2777, nueve de los haplotipos (82%) muestran la configuración 222-314-260 y los dos restantes (18%), la 222-314-264 (
cuadro 2).El alelo de 201 pb del microsatélite D1S2624 está presente en seis de los haplotipos 222-314-260 y en los dos haplotipos 0 222-314-264; los tres haplotipos restantes portan cada uno un alelo distinto para este marcador (203, 207 y 209 pb). En el caso del marcador D1S305, el alelo de 172 pb se encontró en fase con cuatro de los seis cromosomas 222-314-201. En consecuencia, el haplotipo total 222-314-260-201-172 corresponde al 36% (4/11) de los cromosomas con fase. Aquellos cromosomas que portan los alelos 222-314-264-201 portan también el de 162 pb del marcador D1S305; este haplotipo es el segundo más frecuente, encontrado en dos de los once haplotipos (18%). El alelo de 168 pb del marcador D1S305 se halló en dos cromosomas 222-314-260 en fase con distintos alelos del marcador D1S2624, en tanto que los alelos de 170 y 174 pb del primer marcador se encontraron asociados en cromosomas 222-314-260. Finalmente, el haplotipo consenso N370S corresponde a 222-314-260-201-172.
Desequilibrio de ligamiento
Los marcadores con los valores más altos de desequilibrio de ligamiento (d) son 5GC3.2, ITG6.2 y D1S2777. Los alelos de 222 y 318 (pb) de los marcadores 5GC3.2 e ITG6.2 están significativa-mente asociados con la mutación N370S (d = 1, p < 0,01). El único alelo del marcador D1S2777 en desequilibrio de ligamiento significativo con N370S fue el de 260 pb (p < 0,05). Aunque tres de los alelos del marcador D1S305 (162, 168 y 172 pb) obtuvieron valores de d mayores que cero, son bajos y no significativos para la asociación con N370S. Por otro lado, el segundo marcador más distante, D1S2624, presenta un alelo con alta frecuencia en los cromosomas N370S (201 pb), con un valor de d de 0,62 (
cuadro 1).Discusión
El total de microsatélites empleados en el estudio, aunque menor al empleado en otros estudios (8,10), permitió la visualización del comporta-miento del desequilibrio de ligamiento gracias a la distinción de dos zonas dentro de los haplotipos: la fracción conservada y la fracción variable.
Aquellos marcadores incluidos en la fracción conservada son los más cercanos al gen GBA; 5GC3.2 e ITG6.2 están ambos localizados a menos de 10 Kb corriente arriba y abajo del locus GBA, mientras que D1S2777 está localizado aproximadamente a 0,44 Mb corriente arriba del punto de referencia. Teniendo en cuenta estas distancias, menores a 1 centi-Morgan (cM), debe esperarse un número mínimo de entrecruza-mientos entre el locus de referencia y los microsatélites, lo que concuerda con la conservación del haplotipo 222-314-260 en el 82% de los casos.
De igual modo, la existencia de una fracción variable en los haplotipos se explica por la distancia entre el gen GBA y los marcadores D1S2624 y D1S305, localizados a poco más de 1 cM del punto de referencia y con fracciones de recombinación calculadas entre 0,007 y 0,025 (9,10). El proceso de mutación step-wise, común en los micro-satélites, pudo haber contribuido a generar la variación en esta fracción de los haplotipos.
]]> El análisis de desequilibrio de ligamiento es un reflejo de las observaciones hechas en los haplotipos. Aquellos alelos de los microsatélites pertenecientes a la fracción conservada tuvieron los valores de d más altos, en tanto que el desequilibrio de ligamiento decreció hasta casi 0 entre N370S y los alelos de los marcadores de la fracción variable. Sin embargo, en esta fracción, en donde parece encontrarse el límite entre los valores de desequilibrio de ligamiento significativos y los no significativos, el microsatélite D1S2624 presentó un alelo significativamente asociado con N370S, y ninguno de los alelos de D1S305, centromérico con respecto a la mutación, mostró asociación significativa. Esto sugiere la existencia de un factor distinto a la distancia entre loci que determinaría la fuerza de la asociación entre sus alelos; la posición corriente arriba o corriente abajo podría influir sobre este fenómeno, tal como se ha observado en un estudio similar (16).La identificación de un haplotipo consenso N370S (222-314-260-201-172) y la existencia de la relación geográfica entre sus portadores permite suponer que este haplotipo se ha conservado en el tiempo desde que se generó a partir de otro haplotipo ancestral cuyo estado actual hace parte de distintas familias de una misma región cultural y económicamente definida, como lo es el altiplano cundiboyacense. En principio, tal idea tendría total soporte si la configuración de este haplotipo fuera exclusiva de los cromosomas N370S. Como se puede ver en el
cuadro 1, estos alelos, también presentes en los cromosomas control, aparecen con frecuencias que resultan en la inferencia de haplotipos similares en dichos cromosomas al emplear métodos de inferencia no basados en la fase gamética sino en abordajes Bayesianos (17,18) (datos no mostrados). En contraste, los siete haplotipos no N370S inferidos presentan configuraciones distintas, sobre todo en la fracción menor a 1 cM del locus GBA (cuadro 3). Esto confiere una particularidad al haplotipo consenso colombiano N370S; y si bien una misma configuración se observa en algunos de los cromosomas control, ésta puede deberse a que la mutación N370S, al no resultar en un fenotipo grave, no ha sufrido una presión de selección fuerte que resulte en un aislamiento del haplotipo en el que se encuentra respecto del haplotipo que se puede observar en cromosomas normales (19-21).
La configuración y particularidad del haplotipo N370S colombiano, aunque observada en un bajo número de cromosomas, puede ser el resultado de la articulación de procesos demográficos con la ya sugerida ausencia de selección purificante. El principal de estos fenómenos demográficos para efectos de este trabajo comprende los aspectos del origen étnico de la mutación N370S y el camino que ha recorrido desde que se asoció a los alelos que se observan en el actual haplotipo. En este sentido, la comparación del haplotipo consenso colombiano con haplotipos N370S de otras poblaciones puede ayudar a trazar una hipótesis. De las poblaciones ya estudiadas que poseen cierta relación genética e histórica con la colombiana, la española es la más apropiada. Al compararse los haplotipos, se ha visto similitud en uno de los alelos de las fracciones conservadas de ambos grupos de haplotipos, el alelo de 222 pb del microsatélite 5GC3.2 (
cuadro 2). A pesar de que otros estudios basados en el análisis de las frecuencias alélicas de distintos marcadores moleculares en subpolaciones colombianas han logrado demostrar el origen ibérico de algunos de estos marcadores, e incluso de grandes porciones genómicas involucradas (22), la coincidencia de un solo alelo entre haplotipos N370S españoles y colombianos, además de la mutación que define el haplotipo, no proporciona suficiente información para establecer una relación ancestro-descendiente. Por ello se requeriría el estudio de otros marcadores moleculares con la ampliación de los haplotipos, así como el estudio de estos en otras poblaciones relacionadas. Además, si se tiene en cuenta que hasta ahora la mutación Y313H se ha encontrado únicamente en pacientes colombianos y españoles (7,23), la búsqueda de un origen hispánico para algunas de las mutaciones Gaucher, incluida la N370S, es un claro objetivo de estudios posteriores.Agradecimientos
Los autores quieren expresar su agradecimiento a los miembros del Instituto de Errores Innatos del Metabolismo (IEIM), Pontificia Universidad Javeriana, por su amable colaboración en este trabajo. También expresan su gratitud a la "Asociación Gaucher de Colombia" por su ayuda para contactar a los pacientes.
Conflicto de intereses
No existe conflicto de intereses por resultados presentados en este trabajo, y tampoco con las entidades financiadoras.
Financiación
]]> Este trabajo se realizó con fondos de Genzyme Corporation para el estudio de las mutaciones causales de la enfermedad de Gaucher, y de la Pontificia Universidad Javeriana.Correspondencia:
Luis Alejandro Barrera-Avellaneda, Instituto de Errores Innatos del Metabolismo, Pontificia Universidad Javeriana,
Carrera 7 No. 43-82, Edificio Jesús Emilio Ramírez, Bogotá D.C.,Colombia.
Teléfono: (57 1) 320 8320 Ext. 4099 Fax: (57 1) 338 4548
Recibido: 13/02/06; aceptado: 10/07/06
Referencias
1. Beutler E, Grabowski G. Gaucher disease. En: Scriver CR, Beaudet AL, Sly WS Valle D, editors. The metabolic and molecular basis of inherited disease. 7th Edition. New York: Mc Graw-Hill; 2001: 2641-69. [ Links ]
2. Beutler E, Gelbart T. Hematologically important mutations: Gaucher disease. Blood Cells Mol Dis 1998; 24: 2-8. [ Links ]
3. Beutler E, West C, Gelbart T. Polymorphisms in the human glucocerebrosidase gene. Genomics 1992; 12: 795-800. [ Links ]
4. Beutler E, Gelbart T, Kuhl W, Sorge J, West C. Identification of the second common Jewish Gaucher disease mutation makes possible population-based screening for the heterozygous state. Proc Natl Acad Sci USA 1991; 88: 10544-7. [ Links ]
5. Dahl N, Lagerstrom M, Erikson A, Pettersson U. Gaucher disease type III (Norrbottnian type) is caused by a single mutation in exon 10 of the glucocerebrosidase gene. Am J Hum Genet 1990; 47: 275-8. [ Links ]
6. Cormand B, Harboe TL, Gort L, Campoy C, Blanco M, Chamoles N, et al. Mutation analysis of Gaucher disease patients from Argentina: high prevalence of the RecNciI mutation. Am J Med Genet 1998; 80: 343-51. [ Links ]
7. Pomponio RJ, Cabrera-Salazar MA, Echeverri OY, Miller G, Barrera LA. Guacher disease in Colombia: mutation identification and comparison to other Hispanic populations. Mol Genet Metab 2005; 86: 466-72 [ Links ]
8. Rodríguez-Marí A, Díaz-Font A, Chabás A, Pastores GM, Grinberg D, Vilageliu L. New insights in the origin of the Gaucher disease-causing mutation N370S: extended haplotype analysis using the 5GC3.2, 5470 G/A and ITG6.2 polymorphisms. Blood Cells Mol Dis 2001; 27: 950-9. [ Links ]
9. Cormand B, Montfort M, Chabás A, Vilageliu L, Grinberg D. Genetic fine localization of the beta glucocerebrosidase (GBA) and prosaposin (PSAP) genes: implications for Gaucher disease. Hum Genet 1997; 100: 75-9. [ Links ]
10. Díaz GA, Gelb BD, Risch N, Nygaard TG, Frisch A, Cohen IJ, et al. Gaucher disease: the origins of the Ashkenazi Jewish N370 and 84GG acid b-glucosidase mutations. Am J Hum Genet 2000; 66: 1821-32. [ Links ]
11. Cormand B, Grinberg D, Gort L, Chabás A, Vilageliu L. Molecular analysis and clinical findings in the Spanish Gaucher disease population: Putative haplotype of the N370S ancestral chromosome. Hum Mutat 1998; 11: 295-305. [ Links ]
12. Colombo R. Age estimate of the N370S mutation causing Gaucher disease in Ashkenazi Jews and European populations: a reappraisal of haplotype data. Am J Hum Genet 2000; 66: 692-7. [ Links ]
13. Lau EK, Tayebi N, Ingraham LJ, Winfield SL, Koprivica V, Stone DL, et al. Two novel polymorphic sequences in the glucocerebrosidase gene region enhance mutational screening and founder effect studies of patients with Gaucher disease. Hum Genet 1999; 104: 293-300. [ Links ]
14. Centro de Estudios de Polimorfismos Humanos (CEPH)-Fundación Jean Dusset. CEPH Genotype database browser V2.1. Consultado: 13 de julio de 2006. Disponible en:
http://www.cephb.fr/cephdb/php/eng/ [ Links ]15. Risch N, de Leon D, Ozelius L, Kramer P, Almasy L, Singer B, et al. Genetic analysis of idiopathic torsion dystonia in Ashkenazi Jews and their recent descent from a small founder population. Nat Genet 1995; 9: 152-9. [ Links ]
16. Díaz A, Montfort M, Cormand B, Zeng B, Pastores GM, Chabas A, et al. Gaucher disease: the N370S mutation in Ashkenazi Jewish and Spanish patients has a common origin and arose several thousand years ago. Am J Hum Genet 1999; 64: 1233-8. [ Links ]
17. Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 2001; 68: 978-89. [ Links ]
18. Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 2003; 73: 1162-9. [ Links ]
19. Nordborg M, Tavaré S. Linkage disequilibrium: what history has to tell us. Trends Genet 2002; 18: 83-90. [ Links ]
20. Toomajian C, Ajioka RS, Jorde LB, Kushner JP, Kreitman M. A method for detecting recent selection in the human genome from allele age estimates. Genetics 2003; 165: 287-97. [ Links ]
21. Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg TS, Schaffner F, Drake JA, et al. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet 2004; 74: 1111-20. [ Links ]
22. Carvajal-Carmona L, Soto I, Pineda N, Ortíz-Barrientos D, Duque C, Ospina-Duque J, et al. Strong Amerind/White Sex Bias and a Possible Sephardic Contribution among the Founders of a Population in Northwest Colombia. Am J Hum Genet 2000; 67: 1287-95. [ Links ]
23. Cormand B, Vilageliu L, Balcells S, González-Duarte R, Chabás A, Grinberg D. Two novel (1098insA and Y313H) and one rare (R359Q) mutations detected in exon 8 of the b-glucocerebrosidase gene in Gauchers disease patients. Hum Mutat 1996; 7: 272-4. [ Links ] ]]>