ARTÍCULO
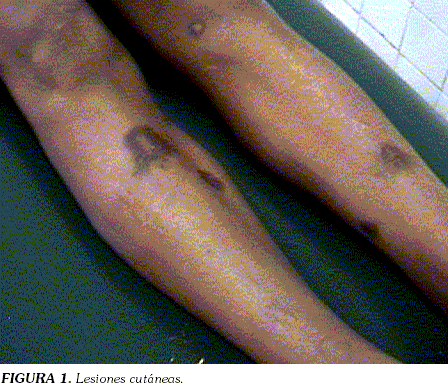
ICTERICIA RECURRENTE EN HOMBRE DE 22 AÑOS. PRESENTACIÓN DE UN CASO DE ESFEROCITOSIS HEREDITARIA A CASE OF HEREDITARY SPHEROCYTOSIS PRESENTING WITH RECURRENT JAUNDICE IN A 22 YEAR-OLD MAN ALEJANDRO PINZÓNa*, FLAVIO VARGASb Y ABNER LOZANOb a Residente Medicina Interna Universidad Surcolombiana,Neiva, Colombia. * Correspondencia: alepyto@yahoo.com. Dirección postal: Dirección postal: Hospital Universitario Hernando Moncaleano Perdomo, Neiva, Colombia. Palabras clave: esferocitosis, fragilidad osmótica, esplenectomía. Abstract Key words: hereditary spherocytosis, osmotic fragility test, splenectomy. Introducción La esferocitosis hereditaria es la anemia hemolítica heredada más frecuente; se caracteriza por que los glóbulos rojos son de forma esférica, debido a alteraciones en las proteínas de membrana que disminuyen su área de superficie (1). La esferocitosis hereditaria afecta a todos los grupos raciales y se estima que la incidencia en caucásicos es de uno por cada 2.000 individuos (1). Su presentación clínica puede ir desde cuadros asintomáticos, hasta crisis hemolíticas severas desencadenadas por procesos infecciosos, que cuando se presentan, pueden complicar el diagnóstico. Aún en formas leves de la enfermedad, las pruebas de fragilidad osmótica se encuentran alteradas. Reporte de caso Hombre de 22 años, remitido al Hospital Universitario de Neiva por cuadro de ocho días de ictericia, dolor en hipocondrio derecho, fiebre, astenia, adinamia, epigastralgia y vómito. Al examen físico: FC: 80xmin, FR: 18xmin, TA: 110/70, T: 35ºC, ictericia marcada, abdomen doloroso a la palpación, con hepatomegalia y esplenomegalia; en extremidades, evidencia de herida en mano derecha y lesiones hiperpigmentadas en miembros inferiores (Figura 1). Como antecedente de importancia, ictericia recurrente desde la niñez en él y en una hermana.
Recibido: Noviembre 21 de 2006. Aceptado: Diciembre 13 de 2006.
b Hospital Universitario Hernando Moncaleano Perdomo, Neiva, Colombia.
Resumen
Durante su segundo día de hospitalización el paciente desarrolló flebitis en miembro superior derecho, fiebre y hematemesis, por lo que requirió de transfusión de hemoderivados, e inicio de tratamiento antibiótico con oxacilina. La endoscopia digestiva alta diagnosticó gastritis eritematosa plana del antro y los hemocultivos se reportaron negativos.
Los resultados de los exámenes de química sanguínea y hemograma se resumen en las Tablas 1 y 2. Otros exámenes de laboratorio fueron: Coombs directo negativo, reticulocitos 6%, hierro sérico: 39 μg/dl (VN 65-175), capacidad total de fijación hierro: 397 μg/dl (VN 250-450), % saturación transferrina: 9,8% (VN 20-50), ferritina sérica: 311 ng/dl (VN 28-397) y albúmina 3,8 g/dl.


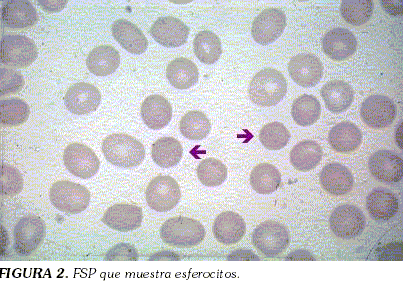
Se realizó aspirado de médula ósea que reportó hiperplasia ligera megacariocítica y eritroide reactiva; el frotis de sangre periférica demostró glóbulos rojos con moderada hipocromia, anisocitosis escasa, con predominio de macrocitos y presencia de esferocitos (+) (Figura 2). Los glóbulos blancos y las plaquetas fueron normales.
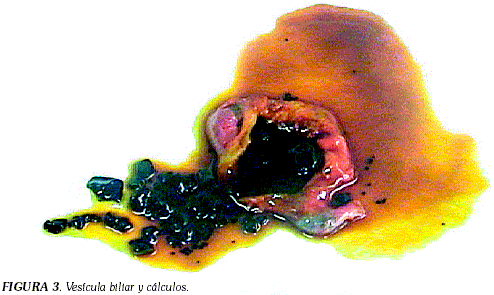
La prueba de fragilidad osmótica se reportó aumentada y la fragilidad osmótica incubada por 24 horas a 37ºC confirmó el resultado (Tabla 3). La flebitis se resolvió y la fiebre desapareció. Se ordenó vacunación con Pneumo23 y se programó para esplenectomia más colecistectomia. Los estudios de patología reportaron la presencia de hiperesplenismo secundario a esferocitosis hereditaria y la vesícula biliar se muestra en la Figura 3.

Discusión
La esferocitosis hereditaria tiene comúnmente una historia familiar, los eritrocitos presentan trastornos en las proteínas de membrana con alteración en la relación volumen/área que los condiciona a sufrir hemólisis esplénica y disminuye su vida en la circulación (2,3). Los casos atípicos pueden requerir de mediciones de proteínas de membrana eritrocitaria y de estudios genéticos para aclarar la naturaleza de la alteración.
Existen mecanismos moleculares que pueden producir esta alteración, el 75% de los afectados exhiben un patrón de herencia autosómica dominante (4), mientras que el 25% restante presentan herencia autosómica recesiva, mutaciones de novo, o carácter dominante con penetrancia incompleta.
Proteínas de membrana eritrocitaria ]]>
Las interacciones de las proteínas de la membrana del eritrocito aseguran su estabilidad y permiten su paso por las fenestraciones de los sinusoides esplénicos. Las interacciones verticales fijan el esqueleto proteico a la doble capa lipídica y las interacciones horizontales brindan estabilidad global a la célula (5).Ankirina: Provee el principal sitio de unión para la espectrina y posee sitios de unión a la membrana celular. Aproximadamente del 15% al 20% de las mutaciones en el gen de la ankirina son mutaciones de novo (6).
Espectrina: Está constituida por subunidades αy β que forman heterodímeros (4, 7).
Banda 3: Constituye el 25% de las proteínas integrales, posee un dominio citoplasmático y otro transmembranoso, para dar estabilidad a la ankirina y a la espectrina.
Proteína 4.2: Estabiliza la interacción entre la ankirina y la banda 3.
Las alteraciones proteicas producen pérdida de lípidos de la membrana eritrocitaria, trastornos metabólicos con aumento en la actividad de la bomba Na/K ATPasa (6) con incremento en la glucólisis y disminución en la concentración de 2,3 difosfoglicerato, lo que produce acidosis intracelular. El pH ácido produce alteración en el cotrasporte K/Cl y finalmente deshidratación celular (8).
La esferocitosis hereditaria puede ser diagnosticada a cualquier edad y su presentación clínica va, desde formas asintomáticas, hasta cuadros de anemia severa que amenazan la vida (4). De acuerdo con el nivel de hemoglobina, bilirrubinas y recuento de reticulocitos, se definen cuatro formas de la enfermedad (Tabla 4).

La forma moderada es la forma más común, del 60% al 75% de los casos; la forma leve se presenta en el 20% y la forma severa en aproximadamente el 5% de los casos (4). ]]>
Característicamente, la ictericia es intermitente y se asocia a hemolisis aumentada por situaciones como exposición al frío, procesos infecciosos, estrés emocional y embarazo. Los extendidos de sangre periférica revelan esferocitos, que en las formas leves de la enfermedad pueden estar ausentes.La esplenomegalia es común, el 75% de los afectados presentan bazo palpable; sin embargo, no parece haber relación entre su tamaño y la severidad de la enfermedad.
Algunas crisis de anemia son precedidas por enfermedades infecciosas bacterianas o virales no específicas, como mononucleosis infecciosa, citomegalovirus, parvovirus B19, etc. (1, 4, 9). Las crisis aplásicas transitorias son causadas por un freno en la eritropoyesis, que se caracteriza por una disminución súbita en la concentración de hemoglobina y reticulocitopenia. En algunas ocasiones, las crisis suceden por el aumento en la síntesis de DNA que agota las reservas de folato. Cuando se presenta mejoría, la recuperación ocurre entre siete y diez días y se caracteriza por aumento en los reticulocitos y trombocitosis.
La litiasis biliar es frecuente (94% a los 13 años), los cálculos son de pigmento biliar (Figura 3) y característicamente se encuentran en jóvenes, durante la segunda década de vida (10, 11).
Algunas manifestaciones poco comunes de la enfermedad son la presencia de úlceras en miembros inferiores (12) y el desarrollo de tumores extra medulares que pueden simular tumores malignos y que sufren metamorfosis grasa luego de que el bazo es removido (13).
Paraclínicos
Los hallazgos clásicos en las pruebas de laboratorio incluyen anemia leve (9-12 g/dl), reticulocitosis, incremento en la concentración media de hemoglobina corpuscular (MCHC > 35) (6, 15), esferocitos en el extendido de sangre periférica, hiperbilirrubinemia y fragilidad osmótica alterada (4).
La hiperbilirrubinemia indirecta es evidente por la destrucción acelerada de los glóbulos rojos, así como la elevación en los niveles de lactato deshidrogenasa en suero; el incremento en MCHC resulta de la deshidratación celular y las pruebas de fragilidad osmótica se alteran, debido a la pobre tolerancia de los eritrocitos a soluciones hipotónicas (6, 13). El 25% de los pacientes pueden tener prueba de fragilidad osmótica normal y su sensibilidad es de aproximadamente el 20% en la esferocitosis leve.
Métodos moleculares
Dentro de los métodos moleculares para establecer niveles de proteínas el más usado es la electroforesis en gel de poliacrilamida (SDS-PAGE) (1). Las alteraciones en las proteínas estructurales de membrana pueden ser individuales o combinadas. ]]>
1. Defecto en espectrina (40%).2. Defecto combinado en espectrina y ankirina.
3. Defecto en banda 3 (20% - 30%).
4. Defecto de proteína 4.2 (1, 3, 7, 16).
La citometría de flujo usando eosin-5-maleimide posee unas altas sensibilidad (92,7%) y especificidad (99,1%) para el diagnóstico de esta enfermedad (4, 17).
Diagnóstico diferencial
Diversas entidades pueden cursar con cuadros similares a la esferocitosis hereditaria, dentro de estas tenemos: incompatibilidad sanguínea ABO; sepsis por Clostridium; reacciones transfusionales; quemaduras extensas; mordeduras de serpientes; picaduras de arañas, abejas o avispas; esplenomegalia por cirrosis o mielofibrosis; anemia hemolítica autoinmune, anemia microangiopática y a talasemia heterocigótica.
Tratamiento
Los pacientes adultos, durante las crisis de anemia, pueden requerir de transfusiones de células rojas y de terapia suplementaria de ácido fólico (4). El uso de eritropoyetina puede ser benéfico y reducir el requerimiento de transfusiones en el primer año de vida (4). La esplenectomía controla las complicaciones de la hemólisis, mejorando la sobrevida de los eritrocitos y actualmente es recomendada para los pacientes con enfermedad moderada a severa, mayores de 6 años (4). En cualquier caso se requiere de vacunación contra Pneumococo, Haemophilus influenzae tipo b y meningococo, previas al procedimiento (4,18). Puede existir ausencia de respuesta a la esplenectomía, siendo necesario descartar la presencia de bazo accesorio, de esplenosis u otras causas de anemia hemolítica.
Algunos autores describen la necesidad de terapia antitrombótica en el peri- operatorio, por el riesgo de trombosis secundaria a la trombocitosis reactiva. La terapia antibiótica profiláctica posterior a la esplenectomía previene el desarrollo de sepsis por gérmenes encapsulados (13). La embolización esplénica y la esplenectomía parcial son alternativas de tratamiento (19, 20). ]]>
La consejería genética es requerida para los estudios familiares y ocasionalmente para descartar otras causas de anemia hemolítica.Referencias
1. Delaunay J. The molecular basis of hereditary red cell membrane disorders. Acta haematologica. 2002;108(4):210-218. [ Links ]
2. Wilkins BS. The spleen. BHJ. 2002;117:265-74. [ Links ]
3. Dulinska I. Stiffness of normal and pathological erythrocytes studied by means of atomic force microscopy. JBBM. 2006;66(1-3):1-11. [ Links ]
4. Bolton-Maggs P. Hereditary spherocytosis; new guidelines. Arch of Disea in Childh. 2004;89(9):809-12. [ Links ]
5. Hassoun H. Hereditary spherocytosis: a review of the clinical and molecular aspects of the disease. Blood. 1996(87):2538-45. [ Links ]
6. Hoffman R. Hematology: Basic Principles and Practice. 4th edition. London: Elsevier Churchill Livingstone; 2005. [ Links ]
7. Gallagher P. Spectrin and ankyrin variants in hereditary spherocytosis. Blood Cells, Molecules, and Diseases. 1998;15:539-43. [ Links ]
8. Greer J. Wintrobe's: Clinical Hematology. 11th edition. London: McGraw Hill; 2003. [ Links ]
9. Bolton-Maggs P. The diagnosis and management of hereditary spherocytosis. Bailliere's Clinical Hematology. 2000;13(3):327-42. [ Links ]
10. Tamary H. High Incidence of early cholelithiasis detected by ultrasonography in children and young adults with hereditary spherocytosis. J Pediatr Hematol Oncol. 2003;25:952-54. [ Links ]
11. Miraglia del Giudice E. Coinheritance of Gilbert Syndrome increases the risk for developing gallstones in patients with hereditary spherocytosis. Blood. 1999:94(7):2259-62. [ Links ]
12. Giraldi S. Leg ulcer in hereditary spherocytosis. Pediatric Dermatology. 2003:20(5):427-28. [ Links ]
13. Beutler E. William's Hematology. 6th ed. New York: McGraw Hill; 2001. [ Links ]
14. Bolton-Maggs P. Guidelines for the diagnosis and management of hereditary spherocytosis. BJH. 2004;126:455-74. [ Links ]
15. Blackman S. Acute Anemia. Clin Ped Emerg Med. 2005;6: 124-37. [ Links ]
16. Hassoun H. Hereditary spherocytosis, with spectrin deficiency due to an unstable truncated ß spectrin. Blood. 1996;87(6):2538-45. [ Links ]
17. Lima P. Band 3Tambau´: a de novo mutation in the AE1 gene associated with hereditary spherocytosis. Eur J Haematol, 2005;74(5):396-401. [ Links ]
18. Fauci A. Harrison's Principles of Internal Medicine, 16th edition. New York: McGraw Hill; 2004. [ Links ]
19. Uranues S. Laparoscopic surgery of the spleen. Surg Clin N Am. 2005;85:75-90. [ Links ]
20. Bader-Meunier B. Long-term evaluation of the beneficial effect of subtotal splenectomy for management of hereditary spherocytosis. Blood. 2001;97: 399-403. [ Links ] ]]>