










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.48 no.3 Bogotá July/Sept. 2023 Epub Apr 08, 2024
https://doi.org/10.36104/amc.2023.2815
Presentación de casos
Linfangioleiomiomatosis esporádica
a Médico Internista. Jefe Servicio de Medicina Interna Clínica Mediláser; Florencia (Colombia).
b Médico Cardiólogo. Servicio de Cardiología; Florencia (Colombia).
c Médicos Generales: Servicio de Medicina Interna. Clínica Mediláser. Florencia (Colombia).
La linfangioleiomiomatosis es una enfermedad rara multisistémica, que afecta principalmente a mujeres en edad reproductiva y tiene como distintivo un compromiso pulmonar quístico difuso. Se puede presentar de forma esporádica o asociada con el complejo de la esclerosis tuberosa. La linfangioleiomiomatosis se caracteriza además por la presencia de angiomiolipomas extrapulmonares, tumores linfáticos, quilotórax y neumotórax.
Se presenta el caso de una paciente de 39 años, que desde su adolescencia viene presentando signos y síntomas de enfermedad pulmonar dados principalmente por tos y disnea progresiva, además de fatiga. Múltiples hospitalizaciones y diversos diagnósticos y tratamientos sin mejoría. Por la historia clínica y la tomografía de alta resolución pulmonar se sospechó la enfermedad, y la determinación del factor de crecimiento endotelial vascular-D contribuyó en el diagnóstico sin nece sidad de biopsia pulmonar. (Acta Med Colomb 2022; 48. DOI:https://doi.org/10.36104/amc.2023.2815).
Palabras clave: linfangiomiomatosis; complejo de esclerosis tuberosa; quistes pulmonares; hipertensión pulmonar; factor de crecimiento endotelial vascular-D
Lymphangioleiomyomatosis is a rare multisystemic disease which mainly affects women of reproductive age and is distinguished by diffuse cystic pulmonary involvement. It may present sporadically or in association with tuberous sclerosis complex. Lymphangioleiomyomatosis is also characterized by the presence of extrapulmonary angiomyolipomas, lymphatic tumors, chylothorax and pneumothorax.
We present the case of a 39-year-old patient who had had signs and symptoms of pulmonary disease since adolescence, predominantly cough and progressive dyspnea, in addition to fatigue. She had multiple hospitalizations and various diagnoses and treatments without improvement. The clinical history and a high-resolution tomography of the lungs led to a suspicion of the disease, and measurement of vascular endothelial growth factor-D contributed to the diagnosis, with no need for a lung biopsy. (Acta Med Colomb 2022; 48. DOI:https://doi.org/10.36104/amc.2023.2815).
Keywords: lymphangioleiomyomatosis; tuberous sclerosis complex; pulmonary cysts; pulmonary hypertension; vascular endothelial growth factor-D
Introducción
El término linfangioleiomiomatosis (LAM) esporádica (LAM-E) se utiliza para pacientes con LAM no asociada con el complejo de esclerosis tuberosa (CET), mientras que LAM-CET hace referencia a LAM que está asociada con el CET. Este raro trastorno pertenece a los tumores de células epitelioides perivasculares, o PEComas, que son neoplasias mesenquimales compuestas por células epitelioides o fusiformes distintivas desde el punto de vista histológico e inmunohistoquímico, y son inmunorreactivas para marcadores de músculo liso y melanocitos 1.
Es frecuente que el diagnóstico de LAM pase desaperci bido y como resultado existe un retardo significativo entre el inicio de la enfermedad y su diagnóstico definitivo. Cualquier enfermedad que manifieste imágenes quísticas pulmonares puede parecerse a una LAM, motivo por el cual la biopsia pulmonar ha sido fundamental para el diagnóstico correcto 2.
En el pasado la biopsia era un enfoque común para diag nosticar LAM, ahora hay un cambio de paradigma hacia métodos menos invasivos que obvian este procedimiento, y especialmente para la LAM-E sin compromiso extrapulmonar como en nuestro caso.
En nuestra paciente el diagnóstico se hizo mediante la historia clínica, las imágenes, y principalmente por la de terminación del factor de crecimiento endotelial vascular-D (FCEV-D). Hasta donde conocemos, es la primera vez que en nuestro país se determina el factor con esta finalidad.
Presentación del caso
Mujer de 39 años procedente del área rural del municipio de Florencia (Caquetá). Casada, no abortos, no hijos, no anti conceptivos, no fumadora, no uso de sustancias psicoactivas. Infección por SARS-CoV-2 en el 2021. Desde los 12 años dificultad para respirar y tos ocasional. Hospitalizaciones en varias oportunidades, sospecha de tuberculosis pero baciloscopias repetidas y prueba de tuberculina no demostrativas, los cultivos fueron negativos. Hospitalizada varias veces en Florencia y en la ciudad de Neiva. Un estudio broncoscópico y análisis del aspirado no fue relevante. Refiere episodios de neumonía y un episodio de neumotórax izquierdo. Nunca biopsia pulmonar. Quedó con diagnóstico de fibrosis pulmo nar de etología a determinar. Utiliza oxigeno domiciliario a 2 L/min.
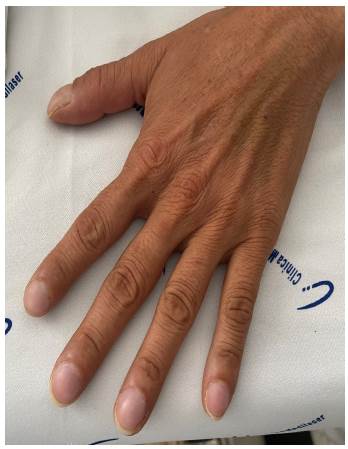
Ingresó con dificultad respiratoria y uso de los músculos intercostales, desaturada (88%, oxígeno 2 L/min), orientada, consciente. Escala de disnea mMRC 4, WHO-FC III, pre sentaba tos y producción de esputo purulento, auscultatoriamente disminución de los ruidos respiratorios y algunos crepitantes difusos, afebril, taquicárdica 124 lpm, signos leves de acropaquía (Figura 1).
Los paraclínicos no mostraron alteraciones incluyendo: anticuerpos antinucleares, anticuerpos nucleares extraíbles, anticuerpos anticitoplasma del neutrófilo, complemento C3 y C4, factor reumatoideo, VIH, Ca 125, VDRL, HBsAg, anti-VHC. Alterados: el recuento de leucocitos: 17 800 por microlitro con neutrofilia 80.8%; proBNP: 1020 pg/mL; hemoglobina y hematocrito 16.3 g/dL y 54.1% respectiva mente. Gases arteriales iniciales: pH 7.31, pCO2 80, pO2 61, HCO3- 40,3, SaO2 88. Cultivo de esputo: pseudomonas aeruginosa multisensible. La espirometría mostró un patrón mixto. No se practicaron: prueba de difusión pulmonar de monóxido de carbono (DLCO), electroforesis de proteínas séricas y cuantificación de cadenas ligeras libres por no estar disponibles en nuestro medio.
Se documentó hipertensión pulmonar por una ecocardiografía sugestiva (VI: rectificación del septum, signo de la D. VD: cavidades dilatadas, dilatación arteria pulmonar de 32 mm, PSAP de 53 mmHg) y se comprobó por cateterismo derecho (presión sistólica arteria pulmonar 52 mmHg, pre sión aurícula derecha de 12 mmHg, en cuña de 11 mmHg, hipoquinesia generalizada y FEVD de 27%).
TAC abdomino-pélvica y de cráneo normales. La to-mografía axial computada (TAC) de tórax se aprecia en la Figura 2.
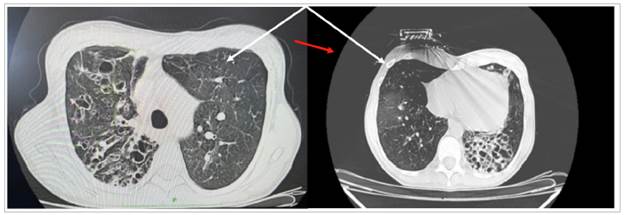
Figura 2 TAC de alta resolución sin contraste. Plano axial. Imágenes quísticas no uniformes en su tamaño. Los quistes suelen ser redondos u ovoides, pero pueden ser poligonales (flecha roja) cuando la afectación del parénquima es grave. Hay engrosamiento septal, tractos fibrocicatriciales, bronquiectasias, nodulos y vidrio esmerilado (flecha blanca).
Ante la sospecha de linfangioleiomiomatosis se solicitó el factor de crecimiento endotelial vascular-D (FCEV-D) a un laboratorio fuera de la ciudad (Synlab). Valor informado de 155.3 pg/mL, (ratificado con el laboratorio), dato no significativo, por lo cual se envió muestra al Laboratorio de Desarrollo y Soporte de Ensayos Traslacionales (TTDSL, siglas en Inglés) del Centro Médico del Hospital Infantil de Cincinnati, Ohio (EE. UU.) en colaboración con la Fundación LAM y teniendo en cuenta el instructivo para el correcto envío de la muestra. Nuevo informe 1533 pg/mL (diagnóstico > 800pg/mL). Se agregó al tratamiento usual sirolimus 1 mg/día.
Discusión
Los signos y síntomas más comunes de la enfermedad son originados por el compromiso pulmonar en la LAM-E. Los hallazgos neurocutáneos (angiofibromas, placas de Shagreen, tubérculos corticales, convulsiones, deterioro cognitivo entre otros) son típicos de la LAM-CET y no se encuentran en la LAM-E 3.
Se desconoce la verdadera incidencia y prevalencia de LAM-E, ya que los datos epidemiológicos disponibles son observacionales y con frecuencia incluyen pacientes con CET. Por consiguiente, la experiencia clínica y la mayoría de los estudios confirman que la variante esporádica de LAM es rara y que afecta casi exclusivamente a mujeres, pero muchas están sin diagnosticar 4. Estimaciones previas sugirieron una tasa de un por millón en la población general; sin embargo, datos más recientes indican tasas más altas, lo que posiblemente refleja los avances en el reconocimiento y diagnóstico de la enfermedad, por ejemplo, The LAM Foun dation estimó una prevalencia de 3-5 por millón de mujeres, pero alrededor de 10-15% de los pacientes registrados en la fundación informan tener CET 5.
La principal anomalía histopatológica en LAM es la proliferación de células atípicas parecidas al músculo liso (células LAM). En el pulmón las células LAM se asocian con múltiples quistes 2.
La disnea durante el ejercicio es el síntoma de presenta ción más común en LAM. Como la disnea es inespecífica y común y la LAM es rara, a menudo se etiqueta a las mujeres con un diagnóstico diferentes antes que se sospeche LAM.
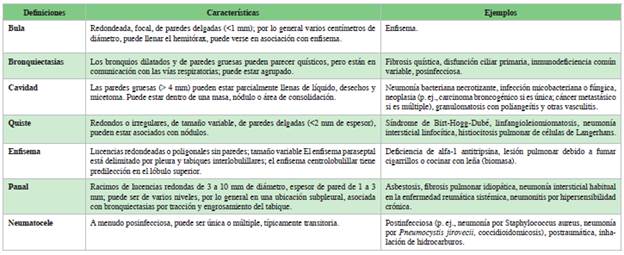
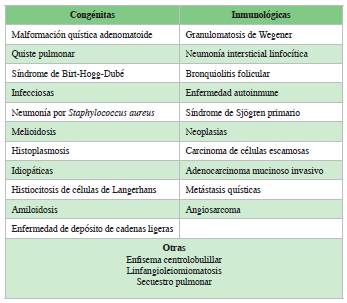
Existe una variedad de enfermedades pulmonares, cuyo hallazgo dominante en la TAC son los quistes. Un quiste es una "transparencia parenquimatosa redonda o un área de baja atenuación con una interfaz bien definida con el pulmón normal" 6. El diagnóstico diferencial de dichas enfermedades se basa en el número, tamaño, forma y distribución de estos, así como en asociación de otros ha llazgos. Los quistes característicos de LAM son de pared delgada, difusos, redondeados, bien definidos, bilaterales y sin predominio lobular. Los quistes suelen medir de 2-5 mm, pero pueden ser más grandes. Pueden ser redondos u ovoides, pero pueden volverse poligonales con afectación parenquimatosa grave. Se han reportado pequeños nódulos centrolobulillares correspondientes a músculo hiperplásico o hiperplasia de neumocitos. Las opacidades focales en vidrio esmerilado pueden deberse a proliferación de músculo liso y la obstrucción linfática puede causar engrosamiento de los tabiques. 6,7. En la serie japonesa, la frecuencia de nódulos pulmonares fue mayor que en los reportes previos 8. En nuestro caso, no encontramos lo típico descrito, existe una mayor desorganización estructural. Aunque la precisión del diagnóstico de LAM basado en la TAC de alta resolución de tórax es alta para los expertos en LAM 7, creemos que basar el diagnóstico solo en la TAC es desaconsejable para la toma de decisiones clínicas. Es posible establecer un diagnóstico clínico confiable de LAM-E cuando el cambio quístico en la TAC es típico de LAM. Pero hay que consi derar que no toda enfermedad quística pulmonar es LAM, pero toda LAM con el tiempo tiene quistes pulmonares. Sin embargo, también hay que ser prudente, porque no toda transparencia parenquimatosa pulmonar son quistes, hay ciertas condiciones que lo simulan (Tabla 1). En la Tabla 2 se muestran algunas de las principales causas de lesiones quísticas pulmonares.
Tabla 1 Transparencia del parénquima pulmonar que simulan quistes. Modificado de la referencia 6 y 7.
Dado el buen rendimiento del biomarcador FCEV-D séri co y tomando el umbral diagnóstico recomendado (>800pg/ mL) por su sensibilidad y especificidad de 73-100% respectivamente, hace que las otras enfermedades pulmonares quísticas, que comúnmente se consideran en el diagnóstico diferencial sean excluidas, porque no elevan el FCEV-D significativamente y evita la biopsia en 70% 9. En los estudios hasta ahora no se han encontrados falsos positivos, pero un resultado negativo no excluye el diagnostico (9,|10).
Es llamativo de este caso:
La TAC de tórax no muestra las imágenes típicas encon tradas en las pacientes, pero a veces lo típico no es lo usual en el mundo real. Consideramos que no debemos descartar la enfermedad sino tenemos las llamadas imá genes típicas y menos sino contamos con un radiólogo experto en quiste pulmonares o LAM. Ante una duda debe solicitarse el biomarcador antes de realizar una biopsia. En nuestro caso, tenemos imágenes atípicas con niveles de FCEV-D típicos desde el punto de vista de sensibilidad y especificidad para LAM 10. Pacientes con LAM-E, como la nuestra, y sin ninguna otra mani festación como angiomiolipomas renales, acumulación de líquido quiloso o CET, establecer un diagnóstico mediante la TAC de alta resolución no es posible, porque la precisión se estima en < 80% en manos de radiólogos expertos 11.
El hallazgo de hipertensión pulmonar grupo 3. Se ha descrito como precapilar, y es generalmente un hallazgo poco frecuente en persona con LAM, ≤ 7%, y cuando se encuentra es de carácter leve 12, contrario a nuestro caso, motivo por el cual se dudó del diagnóstico antes de obtener el FCEV-D. Se planteó la biopsia pulmonar que la paciente rechazó después de conocer los beneficios y riesgo del procedimiento. El compromiso pulmonar se ha descrito como más severo en la LAM-E 12. Es pro bable que la hipertensión pulmonar en nuestra paciente se deba a una vasoconstricción pulmonar hipóxica con remodelación del lecho vascular pulmonar, además del compromiso vascular por fibrosis parenquimatosa in flamatoria debido los procesos infecciosos repetitivos, junto con la afectación bronquial por la proliferación de células LAM y linfáticos, en los cuales la inflamación crónica tiende a complicar la enfermedad. Sin olvidar que los pacientes con LAM muestran una destrucción quística progresiva del parénquima pulmonar.
La acropaquia generalmente no hace parte de los ha llazgos físicos de la LAM 13; sin embargo, en nuestra paciente pudo observarse debido al compromiso cardiopulmonar crónico.
Patrón mixto en la espirometría. Tampoco es lo usual, solo aparece en menos de la cuarta parte de los casos. La mayoría son obstructivo seguido por los normales 13.
Dos resultado de la prueba FCEV-D diferentes. En el primero desconocemos si el embalaje y procesamiento de la muestra fue correcto. Razón por la cual se optó por otro informe en un laboratorio de mayor complejidad y especializado en estos temas ante la persistencia de la sospecha clínica.
Neumotórax a repetición, según distintos autores es característico de LAM con una incidencia entre 61 y 81% 13. Tampoco fue un hallazgo en nuestro caso, por historia solo tiene un episodio de neumotórax.
En conclusión, tenemos a una paciente con LAM-E diagnosticada por los altos niveles de FCEV-D pero que no tiene las características clínicas ni tomográficas usuales informadas en la literatura, por lo tanto, no debe descartarse la enfermedad en una paciente que no tiene lo particulari dades descritas, hasta no agotar las ayudas diagnósticas, y especialmente el biomarcador sérico. En situaciones clínicas apropiadas, la prueba de FCEV-D implica un avance impor tante en el diagnóstico y en su tratamiento actual eficaz, y puede hacer que la biopsia pulmonar sea innecesaria para diagnosticar LAM. Si bien no existe una cura, vivir con la enfermedad es muy diferente hoy que en el pasado, en donde existía temor y miedo ahora hay esperanza, ya no es una sentencia de muerte.
Actualmente lo que existe es una necesidad insatisfecha de biomarcadores adicionales de actividad de la enfermedad y/o respuesta a la terapia. Estos biomarcadores permitirían una atención clínica personalizada y de precisión en el futuro.
Aspectos éticos
Para la publicación se obtuvo el consentimiento informado por parte de la paciente de acuerdo con los principios éticos contenidos en la Declaración de Helsinki, recibió además, aprobación por parte del Comité de Ética de la clínica Mediláser, sucursal Florencia, Colombia.
References
1. Thway K, Fisher C. PEComa: morphology and genetics of a complex tumor fami ly. AnnDiagnPathol. 2015;19(5):359-68. doi: 10.1016/j.anndiagpath.2015.06.003. [ Links ]
2. Kalassian K, Doyle R, Kao P, Ruoss S, Raffin T. Lymphangioleiomyomatosis: new insights. Am J Respir Crit Care Med. 1997;155(4):1183-1186. [ Links ]
3. Matsui K, Tatsuguchi A, Valencia J, Zu-xi Y, Bechtle J, Beasley MB, et al. Extrapulmonary lymphangioleiomyomatosis (LAM): clinicopathologic features in 22 cases. Hum Pathol. 2000; 31(10):1242-8. doi: 10.1053/hupa.2000.18500. [ Links ]
4. Harknett E, Chang W, Byrnes S, Johnson J, Lazor R, Cohen M, et al. Use of variability in national and regional data to estimate the prevalence of lymphangioleiomyomatosis. QJM: An International Journal of Medicine. 2011; 104(11): 971-979. doi.org/10.1093/qjmed/hcr116 [ Links ]
5. The LAM Foundation. https://www.thelamfoundation.org/Newly-Diagnosed/ Learning-About-Lam/About-LAM (Consultado el 8 de noviembre de 2022). [ Links ]
6. Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, Remy J. Fleischner Society: glossary of terms for thoracic imaging. Radiology. 2008;246(3):697-722. doi: 10.1148/radiol.2462070712. [ Links ]
7. Seaman DM, Meyer CA, Gilman MD, McCormack FX. Diffuse cystic lung disease at high-resolution CT. AJR Am J Roentgenol. 2011;196(6):1305-11. doi: 10.2214/AJR.10.4420. [ Links ]
8. Tobino K, Johkoh T, Fujimoto K, Sakai F, Arakawa H, Kurihara M, et al. Computed tomographic features of lymphangioleiomyomatosis: evaluation in 138 patients. Eur J Radiol. 2015;84(3):534-541. doi: 10.1016/j.ejrad.2014.12.008. [ Links ]
9. Young LR, Vandyke R, Gulleman PM, Inoue Y, Brown KK, Schmidt LS, et al. Serum vascular endothelial growth factor-D prospectively distinguishes lymphangioleiomyomatosis from other diseases. Chest. 2010; 138(3):674-81. doi: 10.1378/chest.10-0573. [ Links ]
10. McCormack FX, Gupta N, Finlay GR, Young LR, Taveira-DaSilva AM, Glasgow CG, et al. ATS/JRS Committee on Lymphangioleiomyomatosis. Of ficial American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines: Lymphangioleiomyomatosis Diagnosis and Management. Am J Respir Crit Care Med. 2016; 194(6):748-61. doi: 10.1164/rccm.201607-1384ST. [ Links ]
11. Gupta N, Meraj R, Tanase D, James LE, Seyama K, Lynch DA, et al. Accuracy of chest high-resolution computed tomography in diagnosing diffuse cystic lung diseases. Eur Respir J. 2015;46(4):1196-9. doi: 10.1183/13993003.00570-2015 [ Links ]
12. Freitas CSG, Baldi BG, Jardim C, Araujo MS, Sobral JB, Heiden GI, et al. Pulmonary hypertension in lymphangioleiomyomatosis: prevalence, severity and the role of carbon monoxide diffusion capacity as a screening method. Orphanet J Rare Dis. 2017;12(1):74. doi: 10.1186/s13023-017-0626-0. [ Links ]
13. Ryu JH, Moss J, Beck GJ, Lee JC, Brown KK, Chapman JT, et al. NHLBI LAM Registry Group. The NHLBI lymphangioleiomyomatosis registry: character istics of 230 patients at enrollment. Am J Respir Crit Care Med. 2006;173(1):105-11. doi: 10.1164/rccm.200409-1298OC [ Links ]
Recibido: 21 de Diciembre de 2022; Aprobado: 01 de Abril de 2023
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
