











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
The pharmaceutical industry and academia are current looking for developing effective therapeutic approaches, with fast and safe clinical results [1]. In the pharmaceutical development sector, there is a clear need for more significant incentives in innovation that stimulate the development of health solutions in an accessible way to all society segments. The development of innovative technologies focused on a positive cost-benefit ratio of the available treatments and its security from acquisition to disposal, helps to improve the regulatory tools involved in the available drugs [2]. In this context, the Transdermal Drug Delivery Systems (TDDS) present a challenge in regulatory scenario due to, among others, the technology used for its development that still faces challenges in the regulatory process. The permeation of substances through the skin depends on their physicochemical properties, and their behavior when placed in an appropriate transdermal patch. For this reason, each dosage form must be strictly evaluated in preformulation stages, so that the skin permeation and efficacy studies can be conducted [1].
Currently, to evaluate the efficacy and safety of a transdermal patch, in vitro skin permeation tests are recommended to estimate the in vivo permeation. However, these trials are not present in the main pharmaceutical compendia [3-5]. The in vitro permeation tests take account not only the amount of drug release from the device but also the diffusion capacity of concentration gradient through the skin to a receptor solution, where the amount of permeated drug was determined [6]. On the other side, the release tests evaluate the drug released from its dosage form, so that, it becomes available to be absorbed. The European Pharmacopoeia (EurPh) and the United State Pharmacopoeia (USP) describe this last test [7, 8].
The absence of analytical standards for the permeation test parameters, such as membranes used, apparatus and its dimensions, receiving media, and test time, may impair the in vitro test results. Therefore, it is crucial to analyze the techniques used by the scientific community, in order to guide the development of a harmonized methodology for in vitro permeation test, as well as for the release ones. The standardization of an in vitro permeation methodology is mandatory, not only for the device evaluation in the preformulation stage but also in the quality and safety evaluation of the transdermal delivery systems [9].
Concerning the safety of transdermal patches, the clinical data from currently available devices show that are toxicity risks due to misuse, either by overdose or ingestion of the reservoir gel. Also, there are reports of toxicity related to inappropriate disposal or failure in device adhesion to the skin, where it may accidentally settle on another individual than the patient [10, 11]. In 2005, the Food and Drug Administration (FDA) announced an investigation into deaths and other adverse events resulting from an overdose involving patients using Duragesic®, a transdermal fentanyl device. The device was composed by a reservoir of the drug, a release control membrane, and an adhesive layer to ensure the intimate contact to the skin. Specific reports on failure of the release control membrane, with a subsequent leak of the gel content, led to a product recall in 2004 [12]. Later, technological innovation guided to the development of a device containing the drug in an adhesive matrix, where the adhesive layer promotes intimate contact with the skin and controlled release of the drug [13].
In 2007, the Daytrana®, a transdermal methylphenidate device used on attention deficit and hyperactivity disorder treatment, currently produced by Noven Pharmaceuticals®, had a voluntary recall because of the separation between the device and the protective film. In 2009, the FDA announced a recommendation due to the burn risk when conducting magnetic resonance exams in patients that are using the transdermal devices, that contained a metalized external coating [14]. In 2015, the agency announced again an alert about the loss of skin color associated with the appearance of a chemical leukoderma limited or not to the area of use of the Daytrana® [15].
In the Brazilian context, considering the domestic drug market, the mission of the Brazilian Health Regulatory Agency (Anvisa) is to promote and protect the health of the population and intervene in risks arising from the production and use of medicines and services subject to sanitary surveillance [2]. Besides, judging by the importance of drugs available on the Brazilian market, as well as those that are in the development phase as therapeutic opportunities, the study of the regulatory scenario becomes indispensable in guaranteeing the safety and efficacy of various therapies proposed in transdermal presentations.
Thus, this study aims to compare and analyze the release and permeation test methodologies for transdermal patches described in the main guidance of three regulatory agencies: FDA, EMA, and Anvisa, as well as Organization to Economic Cooperation and Development (OECD) regarding its international importance, reviewing the relevant topics that ensure the quality and safety of these Systems.
METHODS
Regulatory Agencies
An assessment was adopted covering regulatory agencies from three countries or regions: The United States of America (USA), Europe, and Brazil. Respectively, the documents that regulate the scope of TDDS in FDA, EMA and Anvisa were analyzed. Given its importance, the contribution of the Organization to Economic Cooperation and Development (OECD) at the international level was also assessed.
Pharmacopoeias Evaluation
Following the selected countries and regions chosen to regulatory comparison, an approaching regarding the pharmacopeial requirement was made based on the United States Pharmacopoeia (USP), the European Pharmacopoeia (EurPh) and the Brazilian Pharmacopoeia (FB).
RESULTS AND DISCUSSION
Food and Drug Administration (FDA)
One of the most relevant documents that regulate semisolid medicines is the Guidance for Industry: Nonsterile Semisolid Dosage Forms. Scale-up and Postapproval Changes: Chemistry, Manufacturing, and Controls (SUPAC-SS); In vitro Release Testing and In vivo Bioequivalence Documentation. It addresses the requirements to produce nonsterile semisolid pharmaceutical forms in large scale and post-approval changes, covering recommendations to manufacturers about changes in composition, manufacturing process, and increasing or reducing batches of semisolid formulations. It also defines the change levels, the recommended chemical tests for both product in-process and quality control, and the in vitro release test or in vivo bioequivalence tests that support each change level [16].
Specifically, the Draft Guidance for Industry on Transdermal and Topical Delivery Systems: Product Development and Quality Considerations was recently launched to deal precisely with the development and quality of medicines involving topical and trans-dermal delivery systems [17]. The guidance recommends that products need to be developed on the principles of Quality by Design and indicates de use of the Code of Federal Regulation Title 21 (CRF 21) to regulate the quality systems. However, this guidance is still in the implementation phase, not acting as a specific guide for the in vitro release and permeation tests so far, even if it signals what should be implemented soon in the regulatory scenario. It brings relevant recommendations on the tests to be conducted with transdermal patches. The guide proposes to perform characterization studies such as skin permeation, crystallization, thermodynamic stability, system potency, residual drug, in vitro permeation tests, extractable and leachable substances, and heat effects evaluations [17].
In the absence of a specific guide for transdermal patches, the SUPAC-SS has been used. Regarding in vitro tests, the variety of physicochemical tests commonly performed on semisolid products and their components, such as solubility, particle/ droplet size, crystalline form of the drug, viscosity, and homogeneity of the product, historically provided reasonable shreds of evidence of its performance. Thus, it could be considered reasonable to estimate the drug release of the TDDS [16].
Although the guidance mentions that the correlation between in vitro release tests and in vivo expected behavior for semisolid forms is not recommended, the literature presents several studies that support the establishment of an in vitro-in vivo correlation for these products [18-20].
It is important to note that even though the SUPAC-SS, published in 1997, recommends the use of a vertical diffusion cell (Franz diffusion cell) to evaluate the in vitro release, this test was only introduced by USP in 2015. Until the previous year, the dissolution apparatuses 5, 6, and 7 were adopted. Such devices estimate the release of the drug without using a membrane, where the transdermal device should be accommodated in the referred apparatus. Additionally, the position of the TDDS in apparatus 5, for example, is not feasible. The TDDS could be displaced from the disk during the agitation process. Thus, the adhesive face, which should be flat during the test, could bend or even adhere to the walls of the vessel [8, 16].
The use of Franz diffusion cell apparatus allows the permeation and release studies for formulation development, biopharmaceutical characterization, and quality control, both for transdermal patches and semisolid dosage forms. In general, the amount of drug that permeates a skin fixed area during the test could be compared to its potency (EC50) in order to estimate the permeability and check if it is enough to wield the pharmacological effect [21].
According to SUPAC-SS, to regulatory approval, it is necessary that the in vivo test shows bioavailability/bioequivalence of the dosage form [16]. The bioequivalence study project depends on the nature of the drug. Some options include a blank comparative study [22]; comparative clinical experience; or any other validated bioequivalence study, such as a dermato-pharmacokinetic study. The comparative clinical study, usually multicentric, aims to compare the response of a test group that receives a new treatment with that of a control group that receive an existing treatment or a placebo [23]. The dermatopharmacokinetic technique test uses the total of the drug in the stratum corneum and hair follicle as an indication of permeation. The stratum corneum function as a reservoir, and the extent of the drug in this layer could predict the amount absorbed [24]. The recommendations for the bioequivalence and bioavailability tests are still in the implementation phase, available in the Draft Guidance for Industry to Bioequivalence and Bioavailability Studies Submitted in NDAs or INDs.General Considerations, which is broad and not restricted to transdermal patches [25].
Another relevant document from the FDA is the Guidance for Industry to Skin Irritation and Sensitization Testing of Generic Transdermal Drug Product, which advises skin irritation or sensitization tests during the development of transdermal products. As described in the guide, transdermal patches could lead to irritation or sensitization in the skin. In these product developments, the dermatological adverse events are first assessed in animals and then subjected to safe evaluations in large-scale clinical studies. The recommended tests, as well as the methods to be used, are detailed in this guide, and include several tests such as cumulative irritation, sensitization, and combined studies [26]. However, this guide should be updated very soon by the Draft Guidance for Industry to Assessing the Irritation and Sensitization Potential of Transdermal and Topical Delivery Systems for ANDAs, which is still in the implementation phase [27].
As mentioned before, the Guidance for Industry to Transdermal and Topical Delivery Systems: Product Development and Quality Considerations recommends tests to determine the residual drug in transdermal delivery devices should be included in the regulatory approval process. Often, topical, transmucosal, and transdermal dosage forms retain 10 to 95% of the initial amount of the drug after the end of the period of use. This fact leads to a potential safety risk not only for the patient, but also for other family members, caregivers, and pets. For example, adverse events in patients who did not remove the device in the correct period have already been reported and are related to increased or prolonged pharmacological effects. Reports of intoxications and deaths are cited in the scientific literature [10, 11, 13]. Guidance for Industry to Residual Drug in Transdermal and Related Drug Delivery Systems was published in 2011, which supports the need of scientific approach justifications, including safety assessment, based on risks analysis, for the residual drug in the transdermal patches after its period of use, in order to reduce the drug residual levels [28]. The guidance also recommends that a robust product design need to be carried out, using a quality by design approach, as described in the International Council of Harmonisation of Technical Requirements of Pharmaceutical for Human Use Considerations Guideline Q8 (R2) on Pharmaceutical Development and reported in the literature [29, 30].
The amount of absorbed drug in a TDDS depends, among other factors, on the skin contact device area during its use. To the assessment of the adhesive capacity of a TDDS, the Draft Guidance to Assessing Adhesion with Transdermal and Topical Delivery Systems for ANDAs is under development. The document released in 2018 for public consultation provides guidelines for adherence tests, considering that if the TDDS loses adherence during usage, the amount of drug delivered to the patient is reduced, leading to an increased risk of unintended exposure to the medication to another recipient, for example. The guide recommends that adherence studies can be carried out in conjunction with clinical or bioequivalence studies and advises the specifications proposed in the trials as well as the statistical evaluations [31].
European Medicines Agency (EMA)
In 1999, the EMA published the Note for Guidance on Modified Release Oral and Trans-dermal Dosage Forms: Section II. Pharmacokinetic and Clinical Evaluation, including specific recommendations to transdermal systems [32]. In 2014, the Guideline on Quality of Transdermal Patches was published, which proves to be equivalent to the FDA's SUPAC-SS, due to its scope, addressing chemical, manufacturing, physicochemical, and process control criteria. The difference between the documents is that the EMA guideline providing specific recommendations for transdermal devices, that also covers requirements for its particular characteristic, such as adhesive properties, for example [33]. Nonetheless, even it is not mentioned in the SUPAC-SS, the EMA guideline is indicated by USP [5].
The main difference between EMA guideline and SUPAC-SS is that the last suggests, to evaluate the in vitro release, some parameters to be followed using Franz diffusion cells (e.g., diffusion system, temperature, medium receiver), although makes it clear that the manufacturer could look for other references [16] while EMA does not address the Franz diffusion cells and requests that a dissolution development flow should be provided for the product. It also recommends that the TDDS must be evaluated under different receiving medium conditions such as pH, apparatus, agitation, and others. The choosing test must be the most appropriate and discriminative of the in vivo process [33].
Concerning in vitro permeation studies, which are not mentioned by SUPAC-SS, the EMA makes clear that these do not directly correlate with the in vivo process, but are considered a valuable measure for product quality, reflecting the dynamic activity of the drug in the TDDS. The guidance recommends that in vitro permeation studies should be mainly used in development stages of dosage form, and its optimization, not being suitable to the quality control routine. However, it suggests that the in vitro permeation studies could be included in stability study protocols, even at a reduced frequency, to provide product performance data under the indicated storage conditions. The recommendation is that this test should be performed using Franz diffusion cells with a pre-established area where the recipient medium should mimic the in vivo conditions [33].
The adhesive properties evaluations must be conducted by in vitro and in vivo tests. The in vitro test should evaluate the adhesive film removal, and adhesion and removal process from a defined surface, like that reported by USP [5, 33]. In contrast, the in vivo tests should be conducted in the patches proposed period of use, since the product adhesion must be valid so that the clinical trial conclusions could be correctly obtained. For the same reason, the batches used in the clinical trial must be representative of those marketed [33].
The in vivo adhesion study proposed by EMA is quite different from the one proposed by the FDA (Guidance for Industry to Skin Irritation and Sensitization Testing of Generic Transdermal Drug Product) [26]. According to the European agency, the adhesive performance should be included as a component of the clinical studies, or it could be an independent study made with healthy volunteers and patients. If the TDDS has several doses, at least the largest and the smallest devices should be assessed. The assessments elements should include [34]:
Application site.
Protective film residual on the patch or in the skin after a device removal.
The number of transdermal patches attached to the skin.
Cold flow, such as the presence of a dark ring around the device application site during use, move or displacement on the skin, as well as wrinkling.
Device robustness in usual human routine, such as waterproofing on the shower, and saunas, the resistance to moisturizers uses, the removal risk during physical exercises or sleep, and device to close people transferring probability.
Concerning quality control strategies, the manufacturer should establish the pharmacokinetics and clinical efficacy correlations including, in vitro release, in vitro permeation, and in vitro adhesion, whenever possible [34]. The Guideline on Quality of Transdermal Patches also covers the requirements for development and manufacturer of generic drugs, not so different from what is applied to the brand ones [33], and the approach is like the first guidance launched by EMA [32].
Organization for Economic Cooperation and Development (OECD)
The OECD has considerable guidance on dermal and transdermal studies. The main one, published in 2011, is the OECD Guidance Notes on Dermal Absorption (N. ° 156) [35]. This document complements others previously published by this commission in 2004 OECD N. ° 427 and OECD N. ° 428 Guidance, that have been updated recently and address in vivo and in vitro methodologies for absorption studies, respectively, and the OECD Guidance Document for the Conduct of Skin Absorption Studies [35-37]. These guides aim to harmonize the experimental data for dermal and transdermal studies and provide information on alternative methodologies to estimate this parameter. The leading information considers the data type to risk evaluation or estimate relation to public health or pesticide toxicology [35]. From 2004 awards, these guidelines started to be mentioned in the scientific literature that addresses permeation studies. In works conducted by the European Community that approach in vitro permeation methodologies, it is possible to verify references to the OECD guidelines, even though they have no regulatory effects [38-43].
The OECD Guidance for in vitro tests propose that it could be combined with the OECD No. 247 Guidance (in vivo tests) or conducted alone. It also recommends that the OECD Guidance Document for the Conduct of Skin Absorption Studies should be consulted to manage the project based on N.°428. The guidance aims to simplify the choice of appropriate procedures to guarantee the confidentiality of the results [36, 37, 44].
Regarding the extrapolation of results obtained in animals, the literature presents a pertinent observation; In vitro studies performed with rat skin, when well designed and standardized, could predict in vivo absorption. Likewise, using human skin on in vitro assays, it is possible to predict in vivo absorption [43]. The OECD recommends the regulatory approval for in vitro data. However, the guide points out that other regulatory authorities have different acceptance criteria for in vitro methodologies as an estimate of dermal absorption [35]. It is noteworthy that none of the guides published by the OECD refer to release tests.
The in vivo permeation tests have traditionally been used to assess skin absorption for regulatory purposes due to their advantages over in vitro ones, including drug kinetics and dynamic information. However, in vivo tests include the use of live animals, radioactive material and there is, the difficulty determining the initial stages absorption. Besides, there are some differences in the permeability of animal skin and human skin that could harm the test, where the animal skin may overestimate human percutaneous absorption, due to its higher permeability [35].
Among the advantages of in vitro tests, these can be well applied with human skin or other species; the replicate evaluations could be quickly conducted; it does not use live animals; and it is possible to determine the exposure conditions and the damaging impact on skin absorption, avoiding ethical issues. Furthermore, the in vitro method avoids the use of radioactive materials. On the other hand, the in vitro methodology is limited due that peripheral blood flow sink conditions could not be reproduced entirely. However, skin absorption is mainly a passive process, and studies that use appropriate experimental conditions could produce valuable data for several chemical substances, demonstrating the usefulness of this method [35].
The guide also mentions the possibility of data combining from animal and human studies. Notably, this is not a correlation between them, but an approach to use in vivo animals and in vitro human skin data. The methodology is known as the Triple Pack and establishes a relationship among the results to predict human absorption in vivo. The combined data offers a precision result since it corrects the animal results, whose skin permeability is higher than human skin. Meantime, the use of this approach in the regulatory field is still being validated [35, 45, 46].
The EMA guide additionally suggests that national's regulatory authorities may have different acceptance criteria for in vitro absorption studies, and then the choice of which method(s) will be used should be in line with the body regulatory requirements. The only conducting of an in vitro study may be recommended for the first scanning of skin permeation, depending on its intended use. If a more detailed assessment of dermal absorption is required, in vitro and in vivo data should be provided together. The OECD guidance No. 156 also mentions that the most appropriate protocol needs to be verified with the regulatory authority before conducting permeation studies [35]. Although the OECD Guidance 156 is instructive about conducting permeation tests, even drawing a parallel between in vivo and in vitro tests, it is not a consolidated and harmonized protocol yet.
Brazilian Health Regulatory Agency (Anvisa)
In Brazil, Anvisa published GuideN.°20 of February 10th, 2021, postulating the quality requirements to regulatory approval of topic and transdermal products. Until the release of this document, there was no regulation protocol detailing the quality criteria for these dosage forms [47]. The guide is based in international documents previously comment in this study, e.g., the FDA's SUPAC-SS, the EMA Guidance on Quality of Transdermal Patches, and the OECD Guidance Document for the Conduct of Skin Absorption Studies. As well as USP General Chapter 3 "Topical and Transdermal Drug Products - Product Quality Tests, the Guide N.° 20/2021 mentions tests for TDDS quality and safety assurance dividing into general, specific, and transdermal patches assays. The document even shows in vitro performance tests [47].
The general tests include the description of visual changes, such as color, adhesive migration, phase separation, or crystallization. Besides, identification tests, assay, and impurities are also required. The specific tests include uniformity of dosage units; microbial limits, water, and antioxidant content; antimicrobial preservative content and effectiveness; pH; sterility (if applicable); particle/droplet characterization; crystal formation; polymorphism; rheological properties; uniformity in container; and extractable and leachable contents. Regarding the TDDS specific tests, it is the same as those found in the USP, except for static and dynamic shear tests. The tests include the evaluation of the protection film removal, surface adhesion test, immediate adhesion test, flow resistance test, adhesive migration test, and leak test [47].
To in vitro performance devices analysis, the Guide N.° 20/2021 suggests permeation or release tests. The release tests could be performed on the drug's release profile test and should be presented during formulation development. It also includes the comparison with the brand-name drug in case of generic regulatory approval. Those in vitro tests also could be useful in postapproval control changes. To achieve this goal the dissolution apparatus described in official compendia or Franz diffusion cells could be used [47]. The in vitro permeation studies performed in Franz diffusion cells should use human skin or other mammal's membranes. Both permeation and release studies, must be justified while the used methodology and parameters employed.
Concerning the specific requirements for regulatory approval, the TDDS should follow the recommendations of Anvisa (RDC N.°200of December 26 th , 2017). This statute is applied criteria for concession and renew regulatory approval of brand name medicines, generics, and similar. In case of TDDS, both sections for new dosage forms and new drug load ought to have attention. Therefore, for regulatory approval, it is necessary technical justify, safety, and efficacy report containing phase III clinical studies (phase I and II, if applicable) and an appropriate pharmacovigilance project to new formulation/drug load. Concerning the safety of TDDS devices, a risk minimization plan might be necessary [48]. This document aims to manage new risks identified in the postapproval process or even to monitor know risks previously studied. It is also applied in a new therapeutic indication [49]. Thus, the risk minimization plan proves to be consistent with the FDA TDDS safety recommendations in Guidance for Industry to Residual Drug in Transdermal and Related Drug Delivery Systems, which proposes a robust Quality by Design study, in order to ensure the safety and less impact of drug residual generated [28].
In the context of in vitro tests, the Law N.° 11, 794 of October 8th, 2008, as known as Arouca Law, reflects a worldwide trend to promoting scientific and regulatory acceptance of animal-free testing. It launches, as an objective for in vivo tests using animals the same proposed by EMA with the 3 R's (refinement, reduction, and replacement). A significant contribution of this law was the creation of the National Council for the Control of Animal Experimentation (Concea) in Brazil [50]. Normative Resolution N.° 17 ofJuly 3rd, 2014, from Concea, provides validated alternative methods to reduce, replace, or refine the use of animals in research activities. The interested institutions in proposes validating alternative methods must be associated with the National Network on Alternative Methods (Renama), created through Ordinance N.° 491, of July 3rd, 2012, of the Ministry of Science, Technology, and Innovation. The objective of Renama is available through a network of associated laboratories, the methodologies recommended by the OECD, contributing to guarantee the quality of services offered to the productive sector [51, 52].
Additionally, the Normative Resolution (NR) N.° 18 of September 24th, 2014, presents the alternative methods recognized by Concea. There are related seventeen alternative methods grouped by test and all of which are approved by OECD. For in vitro skin permeation tests, the NR indicates the OECD Guide N.° 428 [52]. Therefore, in Brazil, the creation of Concea and Renama are remarkable steps in establishing quality and safety standards for transdermal product development. It is a useful tool, not only to perform pre-clinical trials as for regulatory requirements to in vitro tests in European standards.
Pharmacopoeias Evaluation
U.S. Pharmacopoeia (USP)
According to USP, topical products include, among others, creams, gels, liniments, pastes, suspensions, lotions, foams, sprays, aerosols, solutions, and patches or TDDS. The chapter divides the procedures and acceptability criteria for topical products into universal, specific tests, and specific tests for transdermal delivery systems. These tests are included in General Chapter 3: Topical and Transdermal Drug-Product Quality Tests [5].
Universal Tests for Topical and Transdermal Drug Products
The universal tests for quality attributes are present according to ICH Guidance Q6A Specification-Tests Procedures and Acceptance Criteria for New Drug Substances and New Products: Chemical Substances and includes description, identification, assay, and impurities. The USP redirects the manufacturer to other chapters to guide the specifications of each test. In addition to the universal tests, the specific ones include uniformity of dosage units; microbial limits; antioxidants, water, and antimicrobial preservative content; sterility (if applicable), pH, particle size, crystal formation and in vitro drug release test [5].
A performance evaluation is required in order to guarantee the appropriate product release and evaluate other quality requirements that could affect the drug release from its dosage form. A performance test must be reproducible and reliable. Although it is not an assessment of bioavailability, the test should be able to detect drug release characteristics changes that could modify the expected pharmacological effect. Such changes may be related to the active pharmaceutical ingredient or excipients, physical or physical-chemical properties of the formulation, transport, storage and time conditions, and other critical characteristics for the quality of dosage forms [53].
The TDDS presents a drug-releasing process through passive or active mechanisms. The passive diffusion occurs, exclusively, due to the drug concentration gradient, using or not a permeation agent. On the other hand, the active release mechanism arises using innovative technologies focused in to reduce the stratum corneum barrier effect. Such promising technologies are useful to higher molar mass hydrophilic TDDS drug development. There are some examples in the literature of active TDDS using some techniques such as iontophoresis, sonophoresis, electroporation, and microneedling [54, 55]. The USP, as well as the other compendia, discusses passive TDDS performance tests. These tests need to be developed based on scientific principles so that it could be applied in many drugs development stages, such as research, quality control, equivalence tests, or post-approval changes [8].
According to USP, the TDDS in vitro release test could be conducted using dissolution apparatus 5, 6, or 7. The apparatus 5, also known as Paddle over Disk, is simple and easily applicable to several TDDS devices [8]. Under discussion since 2009 [53], the vertical diffusion cell (Franz diffusion cell) is not included in USP as a performance test yet. However, it is present in the SUPAC-SS [8, 16]. This test wording differs in that the dissolution apparatus is indicated in compliances with a release standard according to individual drug monographs.
On the other hand, the Franz diffusion cell, as previously mentioned, provides information about the TDDS performance. The method is widely used to assess the release and permeation properties of TDDS, as present in the literature [56, 57]. Its evaluation aims to identify the main formulation performance variation that could change the system in vivo bioavailability. The USP 5, 6, and 7 dissolution apparatus are not widely used, while the Franz diffusion cell is often present in scientific methodologies for the drug development stage. So, the mention of the vertical diffusion cell in SUPAC-SS is a remarkable update for manufacturers.
Transdermal Delivery Systems Specific Tests
According to USP, the TDDS is formulated with an adhesive layer in order to ensure intimate skin contact and desired drug dose release. The TDDS adhesive layers used should allow protective film easy removal before use, properly skin adhere during the use, adhesion maintenance for the prescribed time, and easy removal at the end of use, leaving no residue, injuring the skin, or causing any adverse effect. They must also be able to maintain their performance through the product shelf-life. The physical properties that must be tested include peel adhesion, release liner peel, tack, cold flow, shear, and crystal formation [5].
The peel adhesion test measures the force required to remove a TDDS attached to a patterned surface. The temperature and application conditions are previously determined, and then the device is removed with rate and angle remove control. This test should be conducted in three broad categories: peel adhesion test, release liner peel test, and tack test. The in vitro adhesive properties should be characterized according to specification limits determined by the in vivo assays. The acceptance criteria are product specific and define to ensure that the adhesion of each batch is within the range defined by the product design and is consistent between batches based on the product development specifications and statistical evaluation of multiple product batches over the product shelf-life. The cold flow and shear tests measure the cohesive properties of the TDDS and can estimate the flow resistance of the adhesive matrix [5].
In addition to physical tests, for reservoir or pouched TDDS, the leak tests are recommended. This device type must be zero leakage tolerance due to the overdose potential risk. The in-process control methods for leaks or potential leaks requires a development plan from the TDDS manufacturers. The highlight of this approach by USP may evidence the improvement demand of the in-process techniques focused on leak prevention. However, the compendium suggests some tests such as visual inspection, seal integrity, and packaging tests [5].
European Pharmacopoeia (EurPh)
There are no differences between the in vitro tests of EurPh and USP. Instead, it lacks the specific tests for TDDS, such as the adhesion, and the in-process test related by USP. The TDDS in vitro release performance tests also do not include the Franz diffusion cell. The European community compendium indicates the paddle over the disk dissolution apparatus (USP dissolution apparatus 5); the extracting cell, an inert closet apparatus that contains the device applied under a synthetic porous membrane inside the paddle over the disk apparatus; and the cylinders apparatus (USP dissolution apparatus 6) [7].
Brazilian Pharmacopoeia (FB)
The transdermal dosage form is not mentioned in the FB. The recommendations of Guide N.° 20 of February 10 th , 2021, should be followed to assess the in vitro quality control and performance tests [3].
Global overview of the regulatory scenario for transdermal permeation tests
After surveying the regulatory requirements related to TDDS available in the three leading regulatory agencies and OECD, the information about the transdermal dosage forms is summarized in table 1.
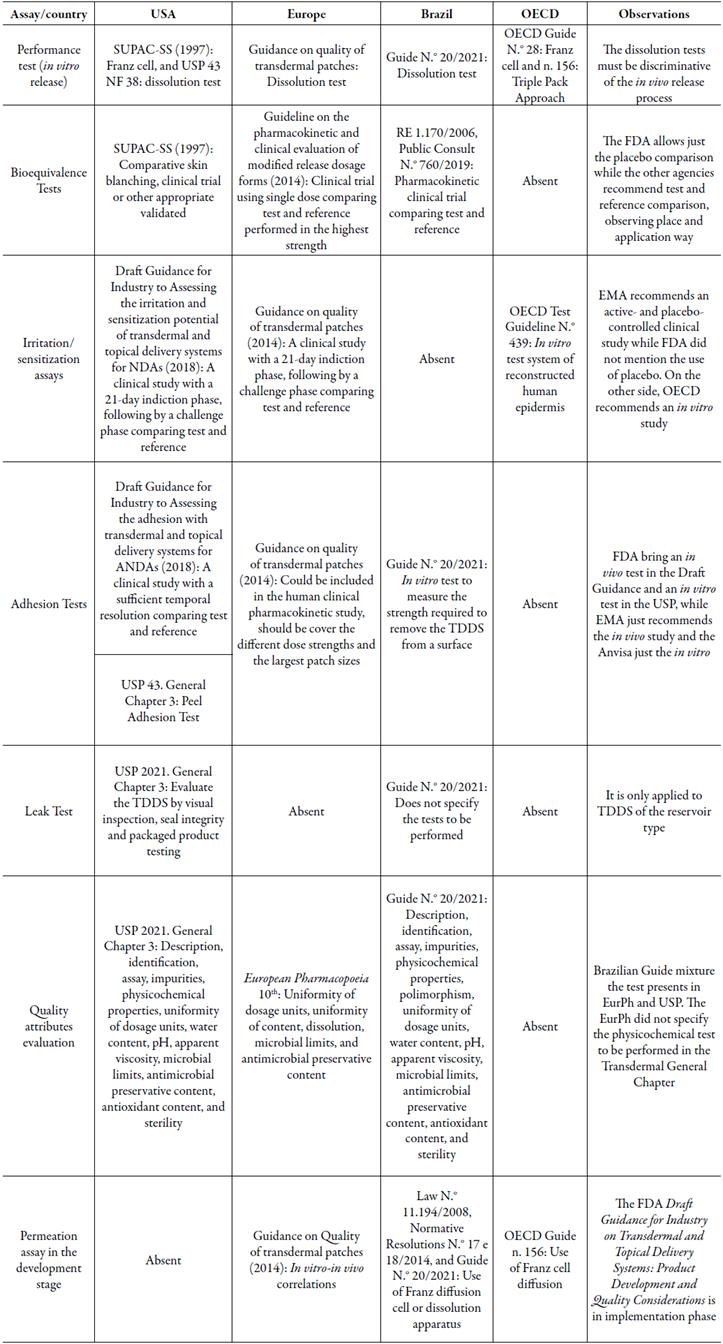
The Brazilian Guide N.° 20/2021, published by Anvisa, brings together important references provided by the FDA, EMA, and OECD that support the national industry on the development of transdermal devices. Despite its launch only in 2019, the RDC N.° 37 of July 6th, 2009, which deals with the foreign pharmacopoeias' admissibility, already allowed the use of USP or EurPh methodologies for TDDS performance, control, and process tests [58]. The launch of the document by Anvisa provides specific guidelines on the Brazilian drug market approval and clarifies the requirements by importers and manufacturers. Likewise, it ensures population safety to transdermal drugs. The document standardizes a roadmap for TDDS trials based on international regulatory agencies.
In the present study, the regulatory requirements for a transdermal drug from three leading agencies were confronted. The FDA and the EMA demonstrate complementarity quality and safety evaluations. The EMA presents a more explicit approach to quality requirements from the TDDS initial development, while the FDA provides extra subsidies in their safety evaluation. The Anvisa took longer to publish a specific guide on this topic. This document mostly follows the international recommendations already established. Regarding the official compendia, USP 2021 presents specific methodologies for the quality control of transdermal devices, an alternative to the lack of approach for these drugs by the Brazilian Pharmacopeia.
CONCLUSION
The number of transdermal formulations has grown in recent decades. The main reason is related to the benefits that this dosage form brings to the treatment of several diseases. It is about a promising methodology when compared with conventional techniques. The in vitro release and permeation tests are crucial for the development and evaluation of the security, efficacy, and performance criteria of these systems. However, there are still discrepancies between the evaluating methodologies by the leading regulatory agencies. Concerning the regulatory approval of new TDDS drug in Brazil, the regulatory scope is still in its first steps and the documents are based on the EMA and FDA primary international documents. The launch of a defined national standardized statute associated with validated in vitro release and permeation tests represents a remarkable breakthrough regarding TDDS.

