










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Colombiana de Psiquiatría
versão impressa ISSN 0034-7450
rev.colomb.psiquiatr. v.29 n.2 Bogotá abr./jun. 2000
DEMENCIA FRONTEMPORAL
UNA REDIMENSIÓN DE LA ENFERMEDAD DE PICK
FRONTOTEMPORAL DEMENTIA
ÓSCAR DOVAL* Y MOISÉS GAVIRIA**
* Servicio de Psiquiatría del Hospital Universitario de Caracas, Venezuela. Grupo Médico HUMANA. Caracas, Venezuela. Neuropsychiatric Fellow -University oí Illinois at Chicago.
**Director Neuropsychiatric División, Department of Psychiatry, University of Illinois at Chicago.
Correspondencia: Osear Doval M.D.
912 S Wood Street, fourth floor, Chicago, Illinois 60612-7327.
E-mail: odoval@hotmail.com.
RESUMEN
La Demencia Frontotemporal, más que una nueva entidad patológica, es la redefinición de la clásica Enfermedad de Pick, como parte de un extenso síndrome con una prevalencia mayor a la imaginada.
Incluye a todos aquellos procesos degenerativos primarios que se inician en las porciones anteriores del cerebro, clínicamente manifiestos por trastornos de conducta y personalidad, más que por alteraciones de carácter cognitivo. En el presente trabajo se hace una revisión bibliográfica del tema, adentrándose en las características clínicas, paraclínicas, neuropatológicas y genéticas.
Palabras clave: Demencia Frontotemporal; Enfermedad de Pick; Neuropatología.
ABSTRACT
Frontotemporal Dementia, is not a new clinical entity but a redefinition of the classical Pick's disease as a part of a extensive syndrome with important and unsuspected prevalence.
Frontotemporal Dementia includes all primary degenerative processes starting in the anterior portions of the brain. This type of dementia is clinically characterized by behavior and personality disorders, more than cognitive alterations. In the present research we performed a bibliographic review of the subject including clinical characteristics, laboratory tests, neuropathology, and genetics.
Key words: Frontotemporal Dementia; Pick's Disease; Neuropathology.
INTRODUCCIÓN
Arnold Pick, entre 1892 y 1906, reportó cuatro casos de pacientes con atrofia del lóbulo frontal y la porción anterior del lóbulo temporal, cuyas manifestaciones clínicas eran básicamente trastornos de conducta y afasia progresiva, lo que fue acuñado como Enfermedad de Pick (1). Este epónimo pasó a ser la representación de un tipo infrecuente, casi exótico de demencia, que ocasionalmente era escuchado como referencia anecdótica ajena. Transcurrió casi un siglo desde estas publicaciones iniciales, hasta que dos grupos de investigadores, en Lund (Suecia) y Manchester (Gran Bretaña), casi de forma simultánea, comenzaron a publicar al final de la década de los 80, una serie de trabajos respecto a un tipo particular de demencia que compromete los lóbulos frontales y la porción anterior de los lóbulos temporales que podía ser definida como entidad clínica particular.
El término de Demencia Fronto-temporal (DFT) es usado para significar todos aquellos procesos degenerativos primarios de la porción anterior del cerebro, caracterizados por sus manifestaciones clínicas, hallazgos en neuroimágenes y elementos histopatológicos, que resultan de especial importancia para la psiquiatría, por su tendencia a presentarse como trastornos de conducta y personalidad; siendo una frecuente causa de demencia en las etapas media y tardía de la vida (2-5).
Le enfermedad ha sido denominada de diversas formas a lo largo de estos últimos años, significando esto, más que un problema semántico, una verdadera fuente de controversia entre los autores, con diversas implicaciones de carácter taxonómico (1). Así, en la literatura podrá encontrarse la misma entidad bajo denominaciones tales como Enfermedad de Pick (EP), Gliosis Subcortical Progresiva (GSP)(6), Demencia Frontal del tipo No-Alzheimer (DFNA)(7), Demencia Frontal (DF)(3), Demencia Frontal sin histología distintiva (DFS-HD) (8-9), Complejo de Desinhibi-ción-Demencia-Parkins onismo^ Amiotrofia (CDDPA)(11) y Demencia Hereditaria con Disfasia-Desin-hibición (DHDD)(11). Por consenso, los grupos de Lund y Manchester decidieron denominarla DFT(12), siendo probablemente el término más extendido y aceptado. Actualmente, en búsqueda de mayor claridad y precisión, la nomenclatura parece evolucionar hacia la de Degeneración Frontotemporal (DgFT)(13).
Aunque la Enfermedad de Alzheimer (EA) continua siendo el tipo de demencia más frecuente en el mundo occidental, con una prevalencia estimada entre 60 y 80% de los casos de demencia(14-15), se ha encontrado que muchos de los pacientes que anteriormente habían recibido el diagnóstico de EA, pueden ser actualmente incluidos dentro de los criterios de DFT.
Diferentes estudios han sugerido que las DFT puede constituir entre el 10 y 20% de los casos de demencia que se inician durante el período presenil de la vida; presentando tan solo entre un 5 y "10% de este grupo, los cambios histológicos característicos de la EP(2,3-5-7-16). En la mayoría de los casos, el inicio de la enfermedad ocurre en la etapa presenil de la vida, con un rango promedio de edad entre los 45 y 65 años y con un discreto predominio en varones, reportándose mayor incidencia familiar que en otros tipos de demencia(3-5-13-21-23).
PRESENTACIÓN CLÍNICA
El inicio de la DFT suele ser insidioso, con progresiva afección de la conducta y deterioro de la personalidad, siendo estos, los principales síntomas. Clínicamente no puede separarse la EP del resto de los tipos de DFT, pudiendo realizarse el diagnóstico diferencial sólo en los estudios postmorten (2-7). Quizás lo más llamativo y de presentación temprana, es la tendencia a la desinhibición, con frecuente rompimiento de normas sociales, falta de tacto, pobre sentido común y capacidad de introspección, impulsividad, ineficiencia e indiferencia ante las actividades laborales y del hogar, descuido del aseo y apariencia personal y eventual agresividad(2,16,24-26). Así mismo, puede observarse conducta de exploración y de utilización, tocando y tratando de usar todos los objetos que encuentran a su paso. También, conducta estereotipada y perseverativa (2-21).
Se presenta además una marcada inquietud con gran distractibilidad, o por el contrario tendencia a la inercia y akinesia(21). La incontinencia urinaria suele ser relativamente precoz (2). Una observación frecuente es el cambio en los hábitos alimenticios, con deseo de comer especialmente alimentos ricos en carbohidratos y disminución de la capacidad de saciedad, lo que conlleva a un incremento de peso. A esta característica particular se la ha llamado "hiperoralidad"; los otros elementos reportados en el síndrome de Klüver Buey (conducta exploratoria con la boca e hipersexua-lidad), resultan menos frecuentes pero se pueden observar(16-27).
El afecto por lo general es aplanado, con falta de espontaneidad, marcada apatía y poca capacidad empática para con familiares y amigos lo cual, en muchas oportunidades, lleva a el aislamiento social. En otros casos, se puede presentar euforia, con marcada e inapropiada jocosidad o humor pueril(2,16-24,28). Es frecuente observar signos clínicos de lo que Blumer y Ben-son originalmente denominaron como tipos de personalidad pseu-dopsicopática y pseudodepresiva del síndrome del lóbulo frontal(24,29,30) como dato más curioso que frecuente, en un estudio reciente se reporta el desarrollo de cualidades artísticas de carácter plástico en pacientes con DFT(31,32).
Han sido descritas manifestaciones psiquiátricas de diversa naturaleza hasta en un 60% de los pacientes. Se destacan: depresión mayor y otros cuadros con humor depresivo hasta en un 15% de los casos; euforia o humor maniforme en un 7%; episodios psicóticos entre un 5 y 7%; y marcada hipocondriasis en el 10%. También se ha reportado trastorno obsesivo-compulsivo de inusual intensidad entre un 6% y 39% de los casos, siendo la conducta de verificación la más observada. Así mismo, puede apreciarse abuso de alcohol con mayor tendencia a accidentes, e incremento del hábito de fumar. No obstante la elevada prevalencia de psicopatología en la DFT, parece ser menos frecuente que en otros tipos de demencia como la EA(35-37).
Al igual que en otros tipos de demencia, se ha encontrado una asociación entre DFT y Enfermedad de Neurona Motora (ENM)(38-44). Usualmente, los signos de deterioro motor siguen al clásico cuadro de alteración conductual ya descrito, más pueden acompañarlo en su inicio e incluso en algunos casos preceder al mismo(43). Generalmente se presenta debilidad muscular, atrofia con signos de parálisis bulbar progresiva, con síntomas poco prominentes de síndrome de neurona motora superior(39-43). De igual manera, se han descrito casos ocasionales de hemiparesia progresiva(45).
Curiosamente, el grueso deterioro nigral, evidenciado en la autopsia, contrasta con la ausencia de notables síntomas extrapiramidales; al parecer, la rápida progresión y la muerte temprana, calculada en un promedio de 3 años tras el inicio de los síntomas motores, impide el cabal desarrollo de los mismos(39,40).
También debe distinguirse, la presencia de síntomas de parkinsonismo en la conocida DFTP-17, que como se mencionará más adelante, puede ser considerada un subgru-po particular de la DFT(46). En ella se observa aplanamiento afectivo, temblor, rigidez, akinesia, trastornos de la marcha y otros tantos signos del cortejo parkinsoniano. Se describen dos grandes formas de presentación ligadas a la misma alteración cromosómica: Demencia con Degeneración Palido-Ponto-Nigral y CDDPA (47).
Más allá de estas manifestaciones neurológicas específicas, en general se podría afirmar que durante las fases tempranas de la DFT el examen neurológico es normal, salvo en algunos casos donde se aprecia presencia de reflejos primitivos como los de "prensión", "succión" y reflejo plantar de extensión. En algunos pacientes se evidencian fasciculaciones, disartria y disfagia. Ya avanzada la enfermedad, el paciente puede mostrar signos extra-piramidales, inmovilidad con akinesia y rigidez, y en raras ocasiones, hasta accesos convulsivos del tipo de las crisis parciales Del mismo modo, ha sido reportada labilidad tensional con tendencia a la hipotensión(2-21).
CRITERIOS CLÍNICOS
De la extensa experiencia de los grupos de Lund y Manchester, surgen por concenso, los criterios para diagnóstico clínico de la DFT. En ellos se sistematiza sus manifestaciones a través de diferentes componentes fenomenológicos, como trastronos de conducta, afectivos, del lenguaje, signos físicos, exámenes paraclínicos, así como las características que respaldan o excluyen el diagnóstico (12). Respecto a estos criterios, han sido publicados estudios que revelan gran agudeza diagnóstica, demostrada por SPECT, previa a la autopsia. No obstante, pueden surgir dudas operativas, ya que no se especifica la cantidad y la jerarquización de criterios para hacer el diagnóstico (ver Tabla 1).

HALLAZGOS
NEUROPSICOLÓGICOS
Algunos autores han encontrado manifestaciones precoces sutiles que fundamentalmente comprometen el área del lenguaje. Sin embargo, para que se hagan evidentes los síntomas cognitivos, suelen transcurrir meses o años(23-50,51). Aunque resulta inusual, también es reportado un discreto compromiso de la memoria, que sólo se detectan con métodos neuropsicológicos especificos(5-17-52). Hasta ya avanzada la afección, estas alteraciones se pueden ver en el marco de una habilidad visoespacial intacta y una función intelectual global normal, lo que permite hacer diagnóstico diferencial con otros tipos de demencia(2-16-33-53). Esto se evidencia en la adecuada realización de pruebas como la Figura Compleja de Rey-Osterrieth y las Matrices Progresivas de Raven(53). Si bien los exámenes neuropsicológicos pueden ser sensibles, resultan poco específicos para diferenciar la DFT de otras afecciones degenerativas que comprometen a las mismas regiones cerebrales (48).
Las alteraciones en el lenguaje se aprecian temprano en la evolución de la enfermedad. Se caracterizan por un discurso fatuo, vacío, carente de espontaneidad y con discreta logopenia; preservándose la articulación, fonología, sintaxis y capacidad de repetición(16-21). También, puede existir anomia leve, donde usualmente está más comprometida la nominación de acciones (dinámica) que la nominación de objetos (estática). Lo anterior, parece ser una función del lóbulo frontal y bien podría servir como parámetro distintivo de la E A (54).
Ya más avanzada la DFT, se acentúa el deterioro verbal con aparición de ecolalia y estenotipia de frases, comentarios y sonidos. Algunos autores consideran esto como una afasia dinámica o transcortical motora, dentro de la clasificación clásica de los síndromes afásicos(16,53,55). Aunque poco comunes, tambien se han descrito, en fases tempranas y medias de la enfermedad, casos de afasia parafásica con fluidez conservada y significante dificultad en la comprensión (afasia de Wernicke), así como de afasia transcortical sensorial (afasia semántica)(2,16,53,55-57). De la misma manera se ha reportado cierta dificultad para apreciar aspectos sutiles del lenguaje, tales como ironía y juego de palabras(58). En su fase tardía, la DFT evoluciona hacia un progresivo mutismo(2-16-21). El deterioro en el lenguaje puede ser evaluado a través de la subescala verbal del WAIS-R, el Examen Diagnóstico para Afasias de Boston, y especialmente los Tests de Nominación Dinámica, que ofrecen la sensibilidad suficiente para detectar los cambios descritos (53).
Aunque tradicionalmente se afirmaba que en la DFT no había compromiso de la memoria, recientemente se han reportado dificultades con las estrategias en el uso de información, más que con el almacenamiento de la misma (2,3-59). La alteración mnésica se hace realmente evidente ya avanzada la enfermedad(52). Se ha informado, en diferentes publicaciones, un compromiso más selectivo de la memoria de trabajo(2-16), del recordar libre y mediante pistas (memoria explícita), así como de la curva de aprendizaje; estos hallazgos parecen orientar hacia un mayor deterioro del hipocampo y en apariencia más característico de la DFT tipo EP(53,60). Menos específicos y poco frecuentes, son casos avanzados con compromiso de la memoria episódica y semántica(16).
Otras áreas cognitivas que pueden resultar afectadas son la atención sostenida, habilidad organizacional (capacidad de planeamiento) y otras funciones ejecutivas, propias(16-21-24) del lóbulo frontal(2-58). Resultan sensibles para estos cambios, pruebas neuropsicológicas tales como las Cartas de Wisconsin, Trail Making Test, Test de Stroop y Tests de Fluidez Verbal; donde usual-mente se puede observar marcado compromiso desde las fases tempranas(3,16-24-58); del mismo modo, parece ser de utilidad el Test de Estimaciones Cognitivas, instrumento diseñado para evidenciar déficit en la capacidad de juicio y razonamiento (61).
Algunos autores, han encontrado que en formas leves de la enfermedad, se observan alteraciones en los tests destinados a detectar dificultad en la toma de decisiones y en el aprendizaje por discriminación visual, presentándose intactos otros paradigmas relativos a disfunción de la corteza prefrontal(62,63). En la fase terminal de la DFT, puede ser vista desorientación espacial y un deterioro cognitivo global, que dificulta enormemente establecer el diagnóstico diferencial con otras entidades que cursan con degeneración cortical(4,30,52,65-67) paramédicos, podría resultar útil el EXIT25, prueba de breve y fácil aplicación, específico para funciones ejecutivas, que tiene una sensibilidad cercana al 100% en pacientes con DFT(64). Otros tests de evaluación cognitiva breve como el MMSE, son pobres indicadores de DFT, ya que arrojan altos porcentajes de falsos negativos(16-64).
ANATOMÍA FUNCIONAL
Los matices clínicos de la DFT tienen una correlación clara con la distribución funcional del proceso degenerativo a lo largo de un eje longitudinal (anteroposterior) y horizontal (izquierdo-derecho). Al comprometerse los dos lóbulos frontales y la porción anterior de los temporales, se observa la clásica clínica de la demencia fronto-temporal, con prominentes cambios de conducta y personalidad(2,16,24,27,68).
Cuando existe distribución asimétrica del proceso degenerativo, con atrofia predominante de las regiones frontotemporales izquierdas, puede emerger un síndrome de Afasia Progresiva Primaria (APP), definida corno una alteración progresiva de la fluidez verbal que conduce al mutismo(69,70-72). Al comprometer el lóbulo temporal izquierdo, se presenta una Demencia Semántica (DS), entendida como pérdida progresiva de los significados de palabras y objetos(70-72). La afección temporal derecha, parece inducir más frecuentemente irritabilidad, impulsividad, alteraciones en la presentación personal, ideas fijas y limitadas, hipomimia e hiperprosexia(73). Una muy rara forma de presentación, es la Proso-pagnosia Progresiva (dificultad en el reconocimiento facial), donde también se ve afectado fundamentalmente el lóbulo temporal derecho(74).
Como es de esperarse, puede también observarse superposición de estos síndromes (69,70-72-75). Así mismo, se ha encontrado emergencia de talento artístico en algunos pacientes con DFT, lo cual aparentemente se debe al deterioro funcional de la región anterior del lóbulo temporal a expensas del hemisferio dominante, preservándose buena parte del lóbulo frontal, especialmente la corteza dorsolateral. La mayoría de estos pacientes muestran habilidades visuales conservadas y mayor compromiso de la función verbal(31,32). Ver figura 1.
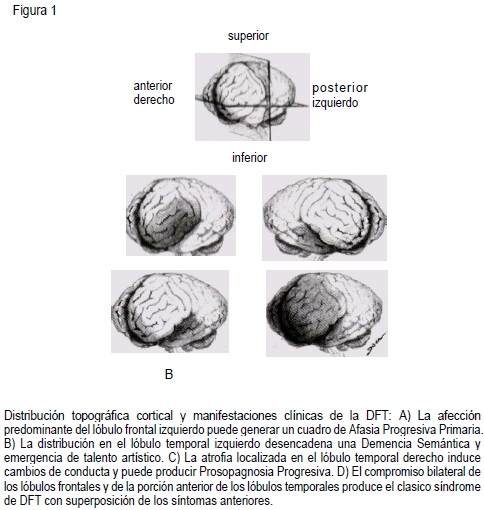
EXÁMENES PARACLÍNICOS
ELECTROENCEFALOGRAFÍA
La mayoría de los estudios sostiene que aún en fases avanzadas de la enfermedad el EEG resulta normal en un elevado porcentaje de casos, lo que indudablemente tendría importancia en el diagnóstico diferencial con otro tipo de demencias, especialmente con la EA, donde resultan típicas las alteraciones electrofisiológicas(1,5,8,12,21,22,52,65,66,76).
Fundamentalmente en pacientes con EP avanzada, al igual que en la EA, se han reportado alteraciones electroencefalográficas, en las que se evidencia disminución de la actividad alfa, ondas lentas difusas y excepcional actividad paroxística con evidencia clinica de convulsiones (48,49). Lejos de lo supuesto, la distribución topográfica de estas alteraciones, no se correlaciona necesariamente con las zonas de atrofia (2). Investigaciones recientes, describen registros electroencefalográ-ficos cuantitativos en DFT con elevada intensidad theta e interacciones sagitales en bandas de alta frecuencia(77).
IMAGEN CEREBRAL
Las imágenes estructurales como la TAC y RMN de cerebro, muestran en la mayoría de los casos atrofia frontal y temporal, con aumento de volumen de los ventrículos laterales y predominio de los cuernos anteriores(2,17,21,48,59,78-82) (figura 2).
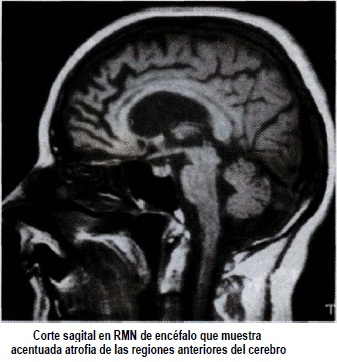
Estos hallazgos son poco específicos y en general imposibilitan la diferenciación con otras enfermedades que cursan con atrofia fron-totemporal, e incluso con la EA que presenta prominente compromiso de las porciones anteriores del cerebro (78,79). Se ha descrito el uso de exposición de superficie hemisférica mediante RMN para valorar el grado y distribución de atrofia presentada en la DFT, encontrándose una significativa localización en regiones anteriores, en Figura 2 comparación con pacientes con EA (83). Una publicación reciente, muestra que la detección del incremento de intensidad de señal por RMN en la sustancia blanca, en fase T2, puede ser un método sensible e incluso específico, que distinge entre DFT y EA(84).
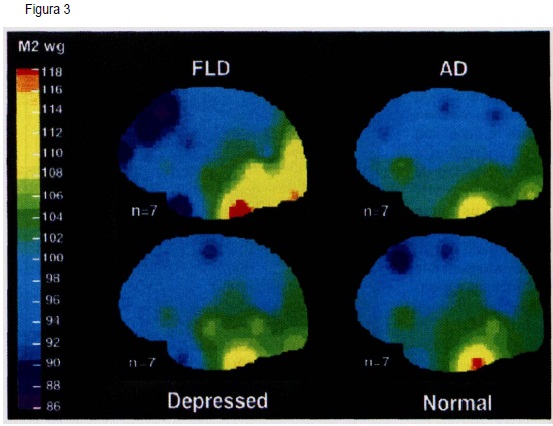
Las imágenes cerebrales funcionales, resultan de especial importancia en la detección de la disminución de metabolismo o flujo sanguineo, que afecta selectivamente las regiones anteriores del cerebro, aún cuando no se evidencian alteraciones en las imágenes estructurales(2,19,48,69,80-82,85-90).
Este patrón, permite hacer una clara distinción entre la DFT, EA, Demencia Vascular (DV), y sujetos sanos (80-86-91). Ver figura 3.
Son realmente escasos los estudios realizados con Espectroscopia Protónica por RMN en DFT; es importante mencionar el trabajo publicado en 1997 por Ernst y colaboradores, donde son analizadas de manera cuidadosa las imágenes por Espectroscopia de Hidrógeno en 14 pacientes con diagnóstico de DFT. En general, se encuentra reducción de los componentes N-Acetil-Aspartato y de Glutamato + Glutamina, ambos sugestivos de pérdida neuronal; asi como incremento del Mioinositol, en aparente relación con aumento del contenido glial. Estos hallazgos fueron especialmente notorios en las regiones frontotemporales y se pudo diferenciar, por estos parámetros, hasta el 92% de los casos con DFT, de los pacientes con EA y sujetos sanos(92).
CURSO Y PRONÓSTICO
Las series de pacientes estudiadas por Gustafson y col., muestran una sobrevida variable de entre 3 y 17 años, con una media de 8,1 años, siendo algo más prolongada (10,5 años) en los casos para el tipo EP (2-21). Este tipo ha sido más estudiado, estableciéndose tres grandes fases, en nuestra opinión no muy diferentes a las observadas en otros tipos de DFT:
- Alteraciones de personalidad y conducta con afección del juicio y trastornos de lenguaje.
- Afasia y deterioro cognitivo con relativa conservación de la memoria y habilidades visoespaciales, y
- Deterioro intelectual global, hipokinesia o akinesia, mutismo y relajación esfinteriana. La muerte usualmente es ocasionada por infecciones de vías urinarias, pulmonares o por úlceras de decúbito(48).
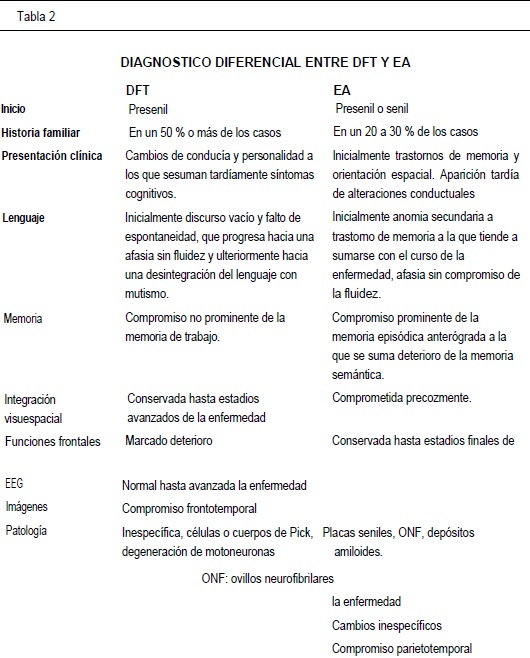
DIAGNÓSTICO DIFERENCIAL
Los cambios iniciales de conducta y personalidad en ausencia de manifestaciones cognitivas ostensibles, sumado a la presencia de co-morbilidad psiquiátrica frecuentemente retarda confunde el diagnóstico de DFT(2-21).
El clásico cuadro clínico de APP se manifiesta como una anomia logo-pénica que puede derivar en un compromiso ulterior de la fluidez verbal, con parafásias tanto semánticas como fonéticas, pudiendo presentarse como diferentes tipos de afasia expresiva (57-94-97), acompañada o no, de otros trastornos cognitivos(56-71-98). La APP está ligada a la DFT, y su diferenciación, si es que cabe, parece estar únicamente determinada por la distribución topográfica de la degeneración cortical, ya que en principio, ambas entidades forman parte de la misma atrofia lobar, presentando quizá, un espectro histopatológico común(7,9,12,56,100,101).
Se ha pensado que la DS podría formar parte del espectro de la DFT, aunque aún resultan poco claras sus características(103) neuropatológicas y etiológicas(2-102). En general, es materia de controversia el estatus nosológico de la relación DFT-ENM. Mientras algunos autores sostienen que se trata de enfermedades independientes e intercurrentes(43,44), otros plantean una interfase entre la DFT y la ENM clásica sin demencia (38,39). Así mismo, se discute la existencia real de esta última entidad, ya que aún cuando no se observan manifestaciones cognitivas, se han descrito alteraciones sutiles en las pruebas neuropsicológicas destinadas a evaluar el lóbulo frontal (39,40). De hecho, actualmente toma fuerza la hipótesis de una superposición genética o al menos neuropatológica y hasta clínica de ambas entidades (9). También, se ha encontrado una asociación entre la Afasia Progresiva Primaria (APP) y la ENM(104).
En la DCB, el diagnóstico diferencial no es complicado clínicamente, ya que se observa akinesia y rigidez progresiva de las extremidades, además de distonía, mioclo-nus, trastornos de la marcha y el equilibrio, apraxia y fenómeno del "miembro ajeno" (105).
Por otra parte, la ausencia de compromiso oculomotor, permite realizar diagnóstico diferencial con la PSP (2-9).
No parece complicada la distinción de la DV, ya que aunque esta se pueda limitar tempranamente a las regiones anteriores del cerebro, el inicio tiende a ser más agudo, la clínica de mayor intensidad y usual-mente las imágenes estructurales y funcionales dan un patrón específico de distribución no uniforme ("parches")(106,107).
Si bien la mayoría de los autores concuerdan en que la EA y la DFT son entidades claramente diferenciables (3-23,24-108), ocasionalmente, los pacientes con EA se presentan con cambios conductuales y compromiso funcional con un predominio frontal detectado por SPECT (87). Aún más, cuando la DFT se encuentra en fase avanzada con severo déficit cognitivo, resulta difícil diferenciarla de la EA w&tww. por el contrario, hay trabajos que aseguran que es posible realizar un diagnóstico diferencial a través de un adecuado análisis fenomenológico retrospectivo, basado en los cambios conductuales y síntomas psiquiátricos, sin tomar en cuenta variables cognitivas(26-12). Incluso, se propone que las diferencias conductuales y cognitivas entre ambas entidades no están directamente relacionadas con la severidad de la demencia, siendo estas estables durante su curso (24).
En las manifestaciones sintomáticas, resulta específico de DFT, la presencia de conducta regresiva e impulsividad, descuido personal, trastornos de la ingesta, conducta perseverativa y reducción de lenguaje (24-113). Por otra parte en la la EA, apenas se aprecia baja motivación y enlentecimiento; estas variables parecen ser tanto o más sensibles que las imágenes funcionales cuando se plantea una diferenciación entre ambas afecciones(24).
En contraste con el curso habitual de la DFT, en la EA se observa un deterioro cognitivo temprano, fundamentalmente de la memoria episódica anterógrada, memoria semántica y de las habilidades visoes-paciales, a las que gradualmente se suman afasia amnéstica, agnosia, apraxia y dificultades en lectoescritura(3-16-114,115). Los síntomas conductuales propios de deterioro frontal, tales como inquietud, agitación, impulsividad y ocasionalmente elementos del síndrome de Klüver-Bucy, se observan en fases avanzadas de la EA (5-16-51). Como claro elemento diferencial con la DFT, las imágenes cerebrales de la EA en su aspecto funcional y estructural, revelan inicialmente, mayor compromiso temporoparietal bilateral, con notable hipoperfusion e hipometabolismo de estas regiones16-117. (Tabla 2)
ANATOMÍA PATOLÓGICA
HALLAZGOS MACROSCÓPICOS
Como cambio inespecífico, se describe atrofia cerebral, la cual resulta más marcada en los lóbulos frontales y la porción anterior de los lóbulos temporales, pero que excepcionalmente puede afectar la porción posterior de estos últimos, así como los lóbulos apriétales, respetando siempre las regiones occipitales(9,18,43,68,119). La atrofia cortícal descrita para el tipo histológico denominado "Degeneración del Lóbulo Frontal", resulta leve y menos circunscrita que para el tipo "EP", donde puede verse marcada atrofia frontotemporal, incluso de la variedad conocida como "filo de cuchillo" (2-21). En la EP ha sido descrita una variante generalizada con extensión del proceso degenerativo al núcleo caudado y que suele comezar a edades más tempranas(48,120) del mismo modo, en la DFT + ENM, se ha descrito menor extensión de la atrofia(68). Típicamente, se presenta dilatación de los ventrículos laterales, particularmente en sus cuernos frontales (10). Aunque inusuales, se ven cambios macroscópicos en los ganglios básales, tálamo, amígdala e hipocampo(2,9,21,68). La ausencia de atrofia del hipocampo, detectada por resonancia magnética volumétrica, podría ser un parámetro que permite diferenciar DFT de EA(121).
HALLAZGOS MICROSCÓPICOS
En general, se presenta importante pérdida neuronal, cambios espongiformes y marcada gliosis, primordialmente distribuidos en las capas superficiales de la corteza frontotemporal(2.9,12,21,68,94,119,122). Estos cambios se observan en especial en la convexidad frontal (en su circunvolución media), lóbulo de la ínsula, región anterior del cíngulo y porción anterior de los lóbulos temporales (3-10).
Hay desacuerdo con respecto al estado del hipocampo, ya que algunos autores proponen degeneración con pérdida neuronal variable, principalmente en las regiones CAÍ y del subiculum(8-119), mientras otros aseguran que esta estructura está casi indemne, tan solo con escasa astrocitosis(3). En casos excepcionales se observa discreto compromiso del cuerpo estriado, núcleos básales, locus cerúleo y porciones posteriores del cerebro (2,8-9).
El estudio celular revela pérdida y estrechamiento neuronal, fundamentalmente a expensas de las células piramidales, pudiéndo estar presentes, como signos de degeneración, estrechas "neuronas fantasmas" y fragmentación neuronal con manchas hipocrómicas (2,9-12). Del mismo modo, se ha documentado compromiso sináptico importante, con reducción cuantitativa(122), además de degeneración esferoidal de los terminales presinápticos(106). Al parecer, la astrocitosis puede ser notable, pudiéndo encontrar correlato en altos los niveles de SlOObeta, una proteína ligada al calcio, encontrada en los astrocitos(106). Cambios del tipo EA están ausentes salvo ocasionalmente en pacientes de edad avanzada(119).
Las células de Pick, presentes específicamente en el tipo homónimo de DFT(9), se observan con mayor frecuencia en hipocampo y amígdala, como células "infladas" o "abalonadas" en proceso de cromatolisis y con núcleo desplazado. Del mismo modo, pueden hallarse inclusiones intracitoplasmáticas densas (Cuerpos de Pick), sin correlación con partículas virales (48). Aunque resulta motivo de controversia, los cuerpos de Pick no son condición sine qua non para el diagnóstico de la enfermedad (123).
Más allá de la distribución topográfica, realmente son escasas las diferencias neuropatológicas entre la DFT y la asociación DFT-ENM, observándose en esta última, cambios más extensos en localizaciones subcorticales (estriatum, tálamo y sustancia nigra). Como es de esperarse, están involucradas las neuronas motoras superiores e inferiores, cuestión que se evidencia por una degeneración en los tractos piramidales medulares. También se ha descrito pérdida de las neuronas motoras en los cuernos anteriores en la médula espinal, así como en el núcleo del hipogloso(9-25,39). Del mismo modo, las células de Betz en la corteza motora están desaparecidas o seriamente estrechadas(39,104). El ENM, se encuentran preservados los núcleos básales de Meynert, el locus cerúleo y el rafe dorsal, y no es posible evidenciar cuerpos de Lewy, de Pick u ovillos neurofibrilares (ONF). Las placas argirofílicas son escasas o están también ausentes(39,56,104).
En la DFTP-17, se ha descrito atrofia de la corteza frontal y temporal, así como de los ganglios básales y sustancia negra. En la mayoría de los casos, estos hallazgos se acompañan de de pérdida neuro-nal y gliosis (46-124). La APP resulta poco específica, puediendo presentar lesiones similares a la DFT con o sin cuerpos de Pick, patología propia de la EA u otros cambios no específicos con o sin inclusiones tipo Enfermedad Difusa por Cuerpos de Lewy, DCB (ws.ioo.iou^ Lo anterior, parece definir la APP como un síndrome clínico, más que como una entidad nosológica particular.
INMUNOQUÍMICA
El área de la inmunohistoquímica promete la posibilidad de reclasifi-car al síndrome aún oscuro de la DFT, arrojando además luces a su etiología y fisiopatología.
En la DFT tipo "Degeneración Frontal" están ausentes los ONF, así como también las placas argirofílicas o están presentes en escasa cantidad, lo que tradicionalmente le ha conferido la nominación: "sin histología distintiva" (9). Más actualmente, se sabe que pueden observarse neuronas corticales "infladas" que resultan aB cristalina reactivas(126,127), además de reacción tau y betatubulina positiva en los esferoides degenerativos observados en algunos terminales presinápticos, lo que parece obligar a una reconsideración de esta denominación inicial(106-128). En la asociación DFT-ENM, se evidencian inclusiones de ubiquitina negativas para tau y aB cristalina, distribuidas en de la corteza frontal, giro dentado del hipocampo, así como, en las células nigrales, neuronas hipoglosales y espinomotoras(12,40,119,129,130).
En la DFTP-17 están presentes ONF-tau positivos localizados en las neuronas y células gliales afectadas, pero con ausencia de depósitos de b amiloide, característica propia de la EA.
En el tipo EP, los cuerpos y células de Pick, son inmunorreactivas para tau, ubiquitina, cromogranina A y plasma B cristalina. Del mismo modo, pueden observarse ONF-tau positivos al igual que en la EA, pero diferiendo de ésta, en la composición de la tau aberrante (55 y 64 kDa doublet)(128-131).
Lo expuesto, permite realizar diagnóstico diferencial entre patología demencial, así los cuerpos y células de Pick, con sus características propias, pueden ser distinguidos de los cuerpos y células de Lewys y de las células infladas de la PSP (132). En la misma línea, como se mencionó anteriormente, la proteína tau aberrante presente en muchas de estas entidades, puede tener características específicas distintivas en la DFTP-17, EA, PSP y Enfermedad Difusa por Cuerpos de Lewys(128,131,2).
CRITERIOS DIAGNÓSTICOS HISTOPATOLÓGICOS
El grupo de Lund-Manchester en 1994, establece sin mayor significación etiológica, tres grandes patrones histológicos descriptivos dentro de la DFT:
- Tipo Degeneración Frontal.
- Tipo EP, y
- Tipo ENM; aclarando que futuros estudios podrían definir un patrón genético que englobe estos tipos como un espectro etiológico, o por el contrario, que cada una de estas manifestaciones histopatoló-gicas llegue a reflejar distintos procesos, gobernados por mecanismos genéticos o moleculares particulares(12) (Tabla 3).

En 1996 el grupo de neuropatología de la Salpetriere (París), propone una forma simplificada de criterios diagnósticos histopatológicos para DFT:
- Si existe una atrofia frontotemporal severa, con marcada pérdida neuronal y astrocitosis, numerosas células infladas y características inclusiones tau y ubiquitina positivas el diagnóstico será de EP.
- Signos de compromiso motor (muchas veces no notados por el clínico) con leve atrofia cortical y escasa espongiosis de las capas II y III, garantizan el diagnóstico de degeneración de DFT-ENM. Las inclusiones ubiquitina positiva pueden ser útiles marcadores específicos.
- Cuando no existen ni inclusiones de Pick, ni ENM, el diagnóstico puede ser DFSHD (95).
ETIOLOGÍA
La mayoría de los estudios en esta área se han desarrollado en el campo genético. Resulta desconcertante la literatura a este respecto, con un espectro que va desde la propuesta de la DFT como una afección esporádica sin una fuerte historia familiar (33-135), hasta autores que asumen de manera más categórica el origen genético del problema, proponiendo además a la DFT como una simple expresión fenotípica de un grupo mayor de patolologías con un mismo sustrato genotípico(13-136). En reportes recientes, se ha documentado que cuando se tienen familiares en primer grado con DFT, se presenta un riesgo hasta 3,5 veces mayor que la población general de padecer esta entidad(137); considerando que la forma familiar de DFT, sería hede-rada siguiendo un patrón autosómico dominante(138).
El hallazgo más consistente en los diversos trabajos, es relación entre alteraciones en el cromosoma 17 y algunos tipos de DFT que cursan con síntomas motores. Dicha relación parece comprometer específicamente al locus 17(21-22), segmento responsable de la síntesis de la proteína microtubular tau(10,48,133,135,136,139-142). Recientemente, se ha denominado este particular subgrupo como Demencia Ponto-temporal con Parkinsonismo ligada al cromosoma 17 (DFTP-17), heredada como entidad autosómica dominante(133) y que se incluye dentro de la interesante clasificación naciente de las taupatologías, caracterizadas por la presencia de inclusiones ricas en formas de fosforilación aberrante de esta proteína <13'46>.
micialmente, los estudios genéticos fueron realizados con cuadros clínicos de diferente denominación, teniendo como elemento común el de tener DFT entre sus manifestaciones patológicas: EP, DFT con Enfermedad de Neurona Motora, (DFT-ENM)(133-134) CDDPA(10-139), DHDD(11). Por otra parte, inclusiones o depósitos de tau pueden observarse también en patologías que típicamente cursan sin DFT como la Esclerosis Lateral Amiotrófica de Guam/Complejo De-mencia-Parkinsonismo, PSP, DCB; existiendo algunas publicaciones que reportan ocasional superposición clínica de estas afecciones con la DFT(136); lo que ha permitido hacer la inferencia de un origen genético común, con diferentes manifestaciones fenotípicas probablemente derivadas de mutaciones dentro del mismo segmento cro-mosómico (13-133). En la misma línea de investigación, se ha descrito recientemente la asociación entre el cromosoma 19, específicamente el locus 19ql3 y la Osteodisplasia Lipomembranosa Poliquística con Leucoencefalopatía Esclerosante; enfermedad hereditaria recesiva de escasa incidencia, caracterizada por quistes óseos sistémicos y DFT presenil progresiva, que ocasiona muerte temprana en las personas que la padecen(144). Así mismo, recientemente se implica una porción del cromosoma 3 (145).
Aunque existen elementos clínicos e histológicos comunes entre la DFT y la enfermedad por priones(146), no se ha encontrado vínculo genético entre estas entidades (147).
Otro aspecto de inusitado interés, es el riesgo de padecer DFT ante la presencia del gen de la apoliproteína E, siendo dicha proteína, como es sabido, un hallazgo propio de la EA <(148,149). Esta proposición resulta aún poco consistente, ya que hay autores que afirman exactamente lo contrario(150).
Al igual que en otras enfermedades degenerativas, se ha pensado que el estrés oxidativo podría jugar un rol en la génesis de la DFT (151).
TRATAMIENTO
Existen pocas publicaciones destinadas específicamente al tratamiento farmacológico de la DFT; hay un reporte de dos casos de DFT con depresión que han respondido a tratamiento con litio y paroxeti-na, probablemente por incremento de la actividad serotoninérgica postsináptica (152). El protocolo con mayor numero de casos hasta ahora publicado, es un estudio abierto con 11 pacientes tratados con ISRS (Fluoxetina, Sertralina o Paroxetina), encontrándose mejoría significativa, al menos en la mitad de los sujetos, de los síntomas depresivos, compulsiones, desinhibición y deseos de consumir carbohidratos(153). Existen estudios realizados con Idaxozan (un agonista selectivo a2 adrenérgico) que muestran importante mejoría de funciones ejecutivas y memoria episódica con incremento del déficit de memoria de trabajo en los pacientes con DFT(154,155).
Basado en el hallazgo de disminución de difosfato de tiamina en la corteza frontal de pacientes con DFT, y en la consabida necesidad de esta sustancia para la síntesis de neurotransmisores, hay autores que proponen emplear tiamina en estudios terapéuticos, pero esto no ha sido hasta el momento explorado(156).
El manejo de las DFT, es pues aun poco claro y esperanzador pues ademas de fármacos dirigidos a mejorar algunas de las manifestaciones psiquiátricas no hay datos acerca de tratamientos específicos para los síntomas demenciales. Se espera que con los estudios de la etiología y la fisiopatología de la en fermedad se desarrollen alternativas terapéuticas que permitan mejorar su pronóstico.
REFERENCIAS
1. Kertesz A. Arnold Pick: A historical introduction. En: Pick's disease and Pick's complex (A Kertesz & D Muñoz eds); Wiley-Liss, NY, 1998:13-21. [ Links ]
2. Lyshman WA: Senile Dementias, Presenile Dementias and Pseudodementias. En: Organic Psychiatry: The Psychological Consecuences of Cerebral Disorder de WA Lyshman. Ed. Blackwell Science, Inc. Third Edition. 1998:428-506. [ Links ]
3. Neary D, Snowden JS, Northen B, Gouldin P. Dementia of frontal lobe type. J Neurol Neurosurg Psychiat 1988;51:353-361. [ Links ]
4. Miller BL, Darby AL, Swartz JR y col. Dietary changes, compulsions and sexual behavior in frontotemporal degeneration. Dementia 1995;6:195-199. [ Links ]
5. Gustafson L. Frontal lobe degeneration of non-Alzheimer type. II Clinical picture and differential diagnosis. Arch Gerontol Geriatr 1987;6:209-223. [ Links ]
6. Neumann MA, Cohn R. Progressive subcortical gliosis, a rare form of presenile dementia. Brain 1967:90:405-418. [ Links ]
7. Brun A. Frontal lobe degeneration of non-Alzheimer type. I. Neuropathology. Arch Gerontol Geriatr 1987;6:193-208. [ Links ]
8. Knopman DS, Mastri AR, Frey WH y col. Dementia lacking distinctive histology features. Neurology 1990:40:251-256. [ Links ]
9. Erisi MM. Other degenerativos diseases causing dementia. En: The neuropathology of dementia de MM Esiri y JH Morris. Cambridge University Press 1997:241-259. [ Links ]
10. Lynch T, Sano M, Marder KS y col. Clinical characteristics of a family with chromosome 17-linked desinhibition-dementia-parkinsonism-am y atrophy complex. Neurology 1994:44:1878-1884. [ Links ]
11. Lendon CL, Lynch T, Norton J y col. Hereditary dysphasic disinhibition dementia. A frontotemporal dementia linked to 17q21-22. Neurology 1998; 50:1546-1555. [ Links ]
12. Brun A, Englund B, Gustafson L y col. Consensus statement. Clinical and neuropathological criteria for frontotemporal dementia. Lund and Manchester Groups. J Neurol Neurosurg Psychiatry 1994;57:416-418. [ Links ]
13. Kertesz A, Muñoz D. Pick's disease, frontotemporal dementia and Pick complex. Emerging concepts. Arch Neurol 1998:55:302-304. [ Links ]
14. Cooper B. The epidemiology of dementia. En: Psychiatry in Elderly de R Jacoby y C Oppenhelmer. Oxford University Press, Oxford 1991: 574-585. [ Links ]
15. Jorn AF, Korten E, Henderson AS. The prevalence of dementia; a quantitative integration of the literature. Acta Psychiatr Scand 1987;76:465-479. [ Links ]
16. Gregory CA, Hodges JR. Clinical features of frontal lobe dementia in comparison to Alzhelmer's disease. J Neural Transm 1996; 47:103-123. [ Links ]
17. Miller BL, Cummings JL, Villanueva-Meyer J y col. Frontal lobe degeneration: Clinical, neuropsychologlcal and SPECT characteristics. Neurology 1991;41:1374-1382. [ Links ]
18. Gregory CA, Hodges JR. Dementia of frontal lobe type and the focal lobar atrophies. Int Rev Psychiatry 1993:5:397-406. [ Links ]
19. Neary D, Snowden JS, Bowen DM y col. Neuropsychologlcal syndromes in presenile dementia due to cerebral atrophy. J Neurol Neurosurg Psychiatry 1986:49:163-164. [ Links ]
20. Neary D, Snowden JS, Mann DMA. The clinical pathological correlates of lobar atrophy. Dementia 1993:4:154-159. [ Links ]
21. Gustafson L, Brun A, Passant U. Frontal lobe degeneration of non-Alzheimer type. En: Unusual Dementlas de MN Rossor BailliéreTindall, London 1992:463-465. [ Links ]
22. Neary D, Snowden JS. Dementia of the frontal lobe type. En Frontal lobe function and dysfunction de HS Levin, HM Eisenberg, AL Sentón. New York, Oxford University Press, 1991:304-317. [ Links ]
23. Gustafson L. Clinical picture of frontal lobe degeneration of non-Alzheimer type. Dementia 1993:4:143-148. [ Links ]
24. Lindau M, Almkvist O, Johansson SE, Wahlund LO. Cognitive and behavioral differentiation of frontal lobe degeneration of the non-Alzheimer type and Alzheimer's disease. Dement Geriatr Cognitive Disord 1998;9:205-213. [ Links ]
25. Pfeffer A, Luczywek E, Golebiowski M, Czyzewski K, Barcikowska M. Frontotemporal dementia: an attempt at clinical characteristics. Dement Geriatr Cogn Disord 1999;10:217-220. [ Links ]
26. Galante E, Muggia S, Spinnler H, Zuffi M. Degenerativo dementia of the frontal type. Clinical evidence from 9 cases. Dement Geriatr Cogn Dlsord 1999;10:28-39 [ Links ]
27. Cummings JL, Duchen LW. Klüver-Bucy syndrome in Pick's disease: clinical and pathological correlations. Neurology 1981;31:1415-1422. [ Links ]
28. Lavenau I, Pasquier F, Lebert F, Petit H, Van der Linden M. Perception of emotion in frontotemporal dementia and Alzheimer disease. Alzheimer Dis Assoc Disord 1999:13:96-101. [ Links ]
29. Blumer D, Benson DF. Personality changes with frontal and temporal lobe lesions. En Psychiatric Aspects of Neurologic Disease de Df Benson y D Blumer: Gruñe & Stratton, New York 1975:151 -170. [ Links ]
30. Miller BL, Chang L, Mena I y col. Progressive right frontotemporal degeneration: clinical, neuropsychological and SPECT characteristics. Dementia 1993: 4:204-213. [ Links ]
31. Miller BL, Cummings J, Mishkin F, y col. Emergence of artistic talent in frontotemporal dementia. Neurology 1998:51:978-982. [ Links ]
32. Miller BL, Pontón M, Benson DF y col. Enhanced artistic creativity with temporal lobe degeneration. Lancet 1996:348:1744-1755. [ Links ]
33. Méndez MF, Selwood A, Mastri AR, Frey WH. Pick's disease versus Alzheimer's disease: a comparison of clinical characteristics. Neurology 1993;43:289-292. [ Links ]
34. Méndez MF, Perryman KM, Miller BLy col. Compulsive behaviors as presenting symptoms of frontotemporal dementia. J Geriatr Psychiatry Neurol 1997; 10:154-157. [ Links ]
35. Berrios GE, Brook P. Delusions and psychopathology of the elderly with dementia. Acta Psychiatr Scand 1985:72:296-301. [ Links ]
36. Burns A, Jacoby R, Levy R. Psychiatric phenomena in Alzheimer's disease. I Disorders of thought content. Br J Psychiatry 1990:157:72-76. [ Links ]
37. Burns A, Jacoby R, Levy R. Psychiatric phenomena in Alzheimer's disease. II Disorders of perception. Br J Psychiatry 1990:157:76-91. [ Links ]
38. Hudson AJ. Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: a review. Brain 1981;104:217-247. [ Links ]
39. Neary D, Snowden JS, Mann DMA, Northen B y col. Frontal lobe dementia and motor neuron disease. J Neurol Neurosurg Psychiat 1990:53:23-32. [ Links ]
40. Talbot PR. Frontal lobe dementia and motor neuron disease. J Neural Transm 1996 47:125-132. [ Links ]
41. Mitsuyama Y, Takamiya S. Presenile dementia with motor neuron disease in Japan. A new entity? Arch Neurol 1979:36:592-593. [ Links ]
42. Mitsuyama Y. Presenile dementia with motor neuron disease in Japan: clinico-pathological review of 26 cases. J Neurol Neurosurg Psychiatry 1984;47:953-959. [ Links ]
43. Mitsuyama Y.Presenile dementia with motor neuron disease. Dementia 1993:4:137-142. [ Links ]
44. Morita K, Kaiya H, Ikeda T, Namba M. Presenile dementia combined with amyotrophy: a review of 34 Japanise cases. Arch Gerontl Geriatr 1987;6:246-263. [ Links ]
45. Schmidtke K, Hiersemenzel LR Progressive hemiparesis in frontal lobe degeneration. Eur Neurol 1997;38:105-112. [ Links ]
46. Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and parkinsonism linked to chromosome 17: a new group oí tauopathies. Brain Pathol 1998:8:387-402. [ Links ]
47. Basum H, Almkvuist O, Axelman K, Brun A y col. Cliniccal characteristics of a chromosome 17-linked rapidly Progressive familial frontotemporal dementia. Arch Neurol 1997:54:539-544. [ Links ]
48. Ure J. Demencias frontotemperales. En: Demencia enfoque multidisciplinario. Eds Mangone CA, Allegri RF, Arizaga RL, Ollari J.A. Ed. Sagitario 1997: 133-142. [ Links ]
49. Cummings JL, Benson DF. Dementia: A clinical approach. Butterworth Publishers 1983:57-69. [ Links ]
50. Benson DF. Progressive froontal dysfunction. Dementia 1993;4:149-153. [ Links ]
51. Brun A, Gustafson L. Psychopathology and frontal lobe involvement in organic dementia. En Alzheimer's disease: Basic Mechanisms, Diagnosis and Therapeutic Strategies de K Iqbal, DRC McLachlan, B Winblad, HM Wisniewsky, Chichester, Wüey 1991:27-33. [ Links ]
52. Ferst H, Besthorn C, Hentschel Fycol. Frontal lobe degeneration and Alzheimer's disease: A controlled study on clinical findings, volumetric brain changes and quantitative electroencephalography data. Dementia 1996;7:27-34. [ Links ]
53. Pillon B, Dubois B, Agid Y. Cognitive deficits in non-Alzheimer's degenerative diseases. J Neural Transm 1996;47:61-67. [ Links ]
54. Cappa SF, Binetti G, Pezzini A y col. Object and action naming in Alzheimer's disease and frontotemporral dementia. Neurology 1998;50:2:351-55. [ Links ]
55. Weintraub S, Rubin N, Mesulam M. Prymary progressive aphasia: longitudinal course, neuropsychological profile and language features. Arch Neurol 1990;47:1329-1335. [ Links ]
56. Kertesz A, Muñoz D. Clinical and pathological of primnary progresive aphassia and frontal dementia. J Neural Trasm 1996;47:133-141. [ Links ]
57. Albert ML, Goodglass H, Helm N y col. Clinical aspects of dysphasia En: Disorders of human comunication 2. Springer-Verlag, New York 1981. [ Links ]
58. Knopman DS, Christensen KJ, Schut LJ y col. The spectrum of imaging and neuropsychological findings in Pick's disease. Neurology 1989;39:362-368 [ Links ]
59. Pasquier F: Neuropsychological features and cognitive assesment in frontotemporal dementia. En Frontotemporal dementia de F Pasquier y P Scheltens. Dordrecht, IGG Publicactions 1996:49-69. [ Links ]
60. Lavenu I, Pasquier F, Lebert F y col. Explicit memory in frontotemporal dementia: The rol of medial temporal atrophy. Dement Geriatr Cogn Disord 1998;9:99-102. [ Links ]
61. Méndez MF; Doss RC, Cherrier MM. Use of the cognitive estimations test to discrimínate frontotemporal dementia from Alzheimer's disease. J Geriatr Psychiatry Neurol 1998;11:2-6. [ Links ]
62. Rahman, Sahakian BJ, Hodges JR, Rogers RD, Robbins TW. Specific cognitive déficits in mild frontal variant frontotemporal dementia. Brain 1999:122:1469-1493. [ Links ]
63. Rahman S, Robbins TW, Sahakian BJ. Comparative cognitive neuropsychological studies of frontal lobe function: implications for therapeutic strategies in frontal variants of frontotemporal dementia. Dement Geriatr Cogn Disord 1999;10[Suppl 1]:15-28. [ Links ]
64. Royall DR, Mahurin RK, Cornell J. Bedside assessment of frontal degeneration: Distinguishing Alzheimer's disease from non-Alzheimer's cortical dementia. Exp Aging Res 1994;20:95-103. [ Links ]
65. Julin P, Wahlund L-O, Basun H y col. Clinical diagnosis of frontal lobe dementia and Alzheimer's disease: Relation to cerebral perfusión, brain atrophy and electroencephalography. Dementia 1995:6:142-147. [ Links ]
66. Levy ML, Miller BL, Cummings JL y col. Alzheimer's disease and frontotemporal dementias. Arch Neurol 1996:53:687-690. [ Links ]
67. Swartz JR, Miller BL, Lesser IM, Darby AL. Frontotemporal dementia: treatment response to serotonin selectíve reuptake inhibitors. J Clin Psychiatry 1997;58:212-216. [ Links ]
68. Mann DMA, South PW. The topographical distribution of brain atrophy in frontal lobe dementia. Acta europathol 1993:85:334-340. [ Links ]
69. Caselli RJ, Jack CR, Petersen RC, Wahner HW, Yanagihara T. Asymetric cortical degenerative syndroms: clinical and radiologic correlations. Neurology 1992;42:1462-1468. [ Links ]
70. Neary D. Frontotemporal degeneraron, Pick disease, and corticobasal degeneration. One entity or 3?. Arch Neurol 1997;54:1425-1427. [ Links ]
71. Mesulam MM. Slowly progressive aphasia without dementia. Ann Neurol 1982:11:592-598. [ Links ]
72. Snowden JS, Neary D, Mann DMA. Frontotemporal lobar degeneration. London, England: Churchill Livingstone Inc: 1996. [ Links ]
73. Edwards-Lee T, Miller BL, Benson DF y col. The temporal lobe variant of frontotemporal dementia. Brain 1997;120:1027-1040. [ Links ]
74. Evans JJ, Heggs AJ, Autoun N, Hodges JR. Progressive prosopagnosia associated with selective right temporal lobeatrophy. Brain 1995:118:1-13. [ Links ]
75. Snowden JS, Griffiths HL, Neary D. Progressive language disorder associated with frontal lobe degeneration. Neurocase 1996;2:429-440. [ Links ]
76. Tissot R, Constantinidis J, Richard J. Picks Disease. En: Handbook of clinical neurology, de JAM Fredericks. Elvieser, Amsterdam 1994:233-246. [ Links ]
77. Besthorn C, Sattel H, Hentschel F, Daniel S y col. Quantitative EEG in frontal lobe dementia. J Neural Transm 1996 47:169-181. [ Links ]
78. Aichner F, Wagner M, Kremser Ch, Felber S. MR-imaging of non-Alzheimer's dementia. J Neural Transm 1996:47:143-153. [ Links ]
79. Aichner FT. Clinical value of magnetic resonance imaging in the diagnosis of demential diseases. Psychiat Danub 1993:5:177-187. [ Links ]
80. Risberg J. Frontal lobe degeneration of non-Alzheimer type III. Regional cerebral blood flow. Arch Gerontol Geriatr 1987:6:225-33. [ Links ]
81. Tyrell PJ, Warrington EK, Frankowiak RSJ, Rossor MN. Progressive degeneration of the right temporal lobe studied with positrón emission tomography. J Neurol Neurosurg Psychiatr 1990;53:1046-1050. [ Links ]
82. Miller BL, Gearhart R. Neuroimaging inthe diagnosis of frontotemporal dementia. Dement Geriatr Cogn Disord 1999;10[suppl 1]:71-74. [ Links ]
83. Kitagaki H, Morí E, Yamaji S y col. Frontotemporal dementia and Alzheimer disease: evaluation of cortical atrphy with automated hemisppheric surface display generated with MR images. Radiology 1998;208:431-439. [ Links ]
84. Kitagaki H, Mori E, Minoro N y col. Alteration of white matter MR signal intensity in frontotemporal dementia. Am J Neuroradiol 1997;18.367-378. [ Links ]
85. Starkstein SE, Migliorelli R, Tesón A y col. Specificity of changes in cerebral blood flow in patients with frontal lobe dementia. J Neurol Neurosurg Psychiatry 1994;57:790-796. [ Links ]
86. Jagust WJ, Reed BR, Seab JP, Kramer JH, Budinger TF. Clinical physiological correlates of Alzheimer's disease and frontal lobe dementia. Am J Physiol Imaging 1989:4:89-96. [ Links ]
87. Neary D, Snowden J, Shields R y col. Single photon emission tomography using 99mTc-HM-PAO in the investigaron of dementia. J Neurol Neurosurg Psychiatry 1987;50:1101-1109. [ Links ]
88. Kumar A, Schapiro MB, Haxby JV y col. Cerebral metabolic and cognitive studies in dementia with frontal lobe behavioral features. J Psychiatr Res 1990:24:97-107. [ Links ]
89. Chase TM. Cortical glucose utilization patterns in primary degenerative dementias of the anterior and posterior types. Arch Gerontol Geriatr 1987:6:289-297. [ Links ]
90. Gregory CA, Serra-Mestres J, Hodges JR. Early diagnosis of the frontal variant of frontotemporal dementia: how sensitive are standart neuroimaging and neuropsychologic tests?. Neuropsychiatry Neuropsychol Behav Neurol 1999:12:128-135. [ Links ]
91. Pickut BA, Saerens J, Marién P y col. Discriminative use of SPECT in frontal lobe-type dementia versus (senile) dementia Alzheimer's type. J Nucí Med 1997;38:929-934. [ Links ]
92. Ernst T, Chang L, Melchor R, Mehringer CM. Frontotemporal dementia and early Alzheimer disease: Differentiation with frontal lobe H-1 MR Spectroscopy. Radiology 1997:203:829-836. [ Links ]
93. Read S, Miller BL, Mena I, y col. SPECT/ pathologic correlations in dementia. J Am Geriatr Soc 1995;43:1243-1247. [ Links ]
94. Carón M. Etude clinique de la maladie Pick. Vigot, París 1934. [ Links ]
95. Hauw JJ, Duyckaerts C, Seilhean D, Camilleri S y col. The neuropathologic diagnostic Criteria of frontal lobe dementia revisited. A estudy of ten consecutive cases. J Neural Transm 1996;47:47-59. [ Links ]
96. Karbe H, Kertesz A, Polk M. Profiles of language imapirment in primary progressive aphasia. Arch Neurol 1993;50:193-201. [ Links ]
97. Miller BL, Cummings JL, Villanueva-Meyer J y col. Frontal lobe degeneratiorvclinical, neuropsychological, and SPECT characteristics. Neurology 1991;41:1374-1382. [ Links ]
98. Wechsler AF. Presenile dementia presenting as aphasia. J Neurol Nuerosurg Psychiatr 1977;40:303-305. [ Links ]
99. Mesulam MM. Primary Progressive Aphasia - Diferentiation from Alzheimer's disease. Ann Neurol 1987:22:533-534. [ Links ]
100. Kertesz A. Frontotemporal degeneration, Pick disease, and corticobasal degeneration. One entity or 3?. Arch Neuroi 1997;54:1427-1429. [ Links ]
101. Lippa CF, Cohén R, Smith TW, Drachman DA. Primary progressive aphasia witth focal neuronal achromasia. Neurology 1991;41:882-886. [ Links ]
102. Hodges JR, Patterson K, Oxbury S, Funnel E. Semantic dementia. Progressive fluent aphasia with temporal lobe atrophy. Brain 1992:115:1783-1806. [ Links ]
103. Snowden J, Griffths H, Neary D. Semantic dementia: autobiographical contribution to preservation of meaning. Cognitive Neuropsychology 1994;11:265-288. [ Links ]
104. Horoupian DS, Katzman R y col. Dementia and motor neuron disease: morphometric bichemical and Golgi studies. Ann Neurol 1984: 16:305-313. [ Links ]
105. Rinne JO, Lee MS, Thompson PD, Marsden CD. Corticobasal degeneration: a clinical study of 36 cases. Brain 1994;117:1183-1196. [ Links ]
106. Zhou L, Miller BL, McDaniel CH y col. Frontotemporal dementia: neuropil espheroids and presynaptic terminal degeneration. Ann Neurol 1998,44:99-109. [ Links ]
107. Sjogren M, Wallin A, Edman A. Symptomatological characteristics distinguish between frontotemporal dementia and vascular dementia with dominant frontal lobe syndrome. In J Geriattr Psychiatry 1997;12.656-661. [ Links ]
108. Souliez L, Pasquier F, Lebert F y col. Generation effect in short term verbal and visuspatial memory: comparisons between dementia of Alzheimer type and dementia of frontal lobe type. Cortex 1996:32:347-356. [ Links ]
109. Jagust WJ, Budinger TF, Reed BR. The diagnosis of dementia with single photon emission tomography. Arch Neurol 1987:44:258-262. [ Links ]
110. Méndez MF, Perrymar KM, Miller BL, Cummings JL. Behavioral diffrences between frontotemporal dementia and Alzheimer's disease: A comparison on the BEHAVE-AD rating scale. Int Psychogeriatr 1998:10:155-162. [ Links ]
111. Swartz JR, Miller BL, Lesser IM y col. Behavioral phenomenology in Alzheimer's disease, frontotemporal dementia, and late-life depression: a retrsopective analysis. J Geriatr Psychiatry neurol 1997;10:67-74. [ Links ]
112. Gregory CA: Orrell M; Sahakian B, Hodges J. Can frontotemporal dementia and Alzheimer's disease be differenciated using a brief battery of tests?. Int J Geriatr Psychiatry 1997;12:375-383. [ Links ]
113. Miller BL, Ikonte C, Pontón M y col. A study of the Lund-Manchester criteria for frontotemporal dementia: clinical and single-photon emission CT correlations. Neurology 1997:48:937-942. [ Links ]
114. Greene JDW, Baddeley AD, Hodges JR. Autobiographical memory and executive function in early dementia of Alzheimer type. Neuropsychologia 1995:33:1647-1670. [ Links ]
115. Hodges JR, Patterson K. Is semantic memory consistently impaired early in the course of Alzheimer's disease? Neuroanatomical and diagnostic implications. Neuropsychologia 1995:33:441-459. [ Links ]
116. Klatka LA, Schiffer RB, Powers JM, Kazee AM. Incorret diagnosis of Alzheimer's disease. Arch Neurol 1996;53:35-42. [ Links ]
117. Chang L, Yener GG, Miller BL, Mehringer M. Magnetic resonance spectroscopy and single photon emission tomography in Alzheimer's disease: New directions. Facts Res Gerontol 1994;5:67-78 [ Links ]
118. Mazziotta JC, Frackowiak RSJ, Pheips ME. The use of positrón emission tomography in the clinical assesment of dementia. Sem Nucí Med 1992;12:233-246. [ Links ]
119. Mann DMA, South PW, Snowden JS, Neary D. Dementia of frontal lobe type: neuropathology and inmunochemistry. J Neurol Neurosurg Psychiat 1993;56:605-614. [ Links ]
120. Muñoz García D, Ludwin SK. Classic and generalized variants of Pick's disease: A clinicopathological, ultrastructural, and inmunocitochemical comparative study. Ann Neurol 1984;16:467-480. [ Links ]
121. Frisoni GB, Laakso MR Beltramello A, Geroldi C, Bianchetti A, Soininen H, Trabucchi M. Hippocampal and entorhinal cortex atrophy in frontotemporal dementia and Alzheeimer's disease. Neurology 1999:52:91-100 [ Links ]
122. Brun A, Liu X, Erikson C. Synapse loss and gliosis in the molecular layer of the cerebral cortex in Alzheimer's disease and frontal lobe degeneration. Neurodegeneration 1995;4:171-177. [ Links ]
123. Schmitt HP, Yang Y, Forstl H. Frontal lobe degeneration of non-Alzheimer type and Pick's atrophy lumping or splitting? Eur Arch Psychiatry Clin Neurosci 1995:245:299-305. [ Links ]
124. Hulette CM, Pericak-Vance MA, Roses AD, Schmechel DE, Yamaoka LH, Gaskell PC, et al. Neuropathological features of frontotemporal dementia and parkinsonism linked to a chromosome 17q21-22 (FTDP-17): Duke family 1684. J Neuropathol Exp Neurol 1999;58:859-866 [ Links ]
125. Turner RS, Kenyon LC, Trojanowski JQ, Gonatas N, Grossman M. Clinical neuroimaging, and pathologic features of progressive nonfluent aphasia. Arch Neurol 1996:39:166-173. [ Links ]
126. Cooper PN, Jackson M, Lennox G y col. Tau, ubiquitin and alpha B crystalline ¡nmunochemistry define the principal causes of degenerative frontotemporal dementia. Arch Neurol 1995:52:1011-1016. [ Links ]
127. Jackson M, Lowe J. The new neuropathology of degenerative frontotemporal dementias. Acta Neuropathol 1996;91:127-134. [ Links ]
128. Jellinger KA. Structural basis of dementia in neurodegenerative disorders. J NeuralTransm 1996;47:1-29. [ Links ]
129. Okamoto K, Murakami N, Yoshida H, Hashizume M y col. Ubiquitin-positive intraneuronal inclusions in the extramotor cortices of presenile dementia patients with motor neuron disease. J Neurol 1992;239:426-430. [ Links ]
130. Lowe J. new pathological findings in amyotrophic lateral sclerosis. J Neurol 1994:124:38-51. [ Links ]
131. Snowden JS, Neary D, Mann DW y col. Progressive language disorder due to lobar atrophy. Ann Neurol 1992;31:174-183. [ Links ]
132. Dickson DW, Feany MB, En SH, Mattiace LA, Davies P. Cytoskeletal pathology in non-Alzheimer degenerative dementia: new lesions in diffuse Lewy body disease, Pick's disease, and Corticobasal degeneration. J Neural Transm 1996:[Suppl 1]47:31-46. [ Links ]
133. Poorkaj R Bird T, Wijsman E, y col. Tau Is a Candidate Gene for Chromosome 17 Frontotemporal Dementia. Ann Neurol 1998:43:815-825 [ Links ]
134. Chang L, Cornford M, Miller BL y col. Neuronal ultrastructural abnormalities in a patíent with frontotemporal dementia and amyotrophic lateral sclerosis. Dementia 1995;6:1-8. [ Links ]
135. Wihelmsen KC, Clark LN, Miiler BL, Geschwind DH. Tau mutations in frontotemporal dementia. Dement Geriatr Cogn Disord 1999;10[suppl1]:88-92. [ Links ]
136. Wilhelmsen KC. Frontotemporal dementia is on de MAPt. Ann Neurol. 1997;41:139-140. [ Links ]
137. Stevens M, van Dujin CM, Kanphorst W, y col. Familiar aggregation frontotemporal dementia. Neurology 1998:50:1541-1545. [ Links ]
138. Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol 1999:56:817-822. [ Links ]
139. Bird TD. Genotypes, phenotypes and frontotemporal dementia. Take your Pick. Neurology 1988:50:1526-1527. [ Links ]
140. Foster NL, Wilhelmsen K, Sima AAF y col. Frontotemporal dementia and parkinsonism línked to chromosome 17: a consensus conference. Ann Neurol 1997;41:706-715. [ Links ]
141. Bird TD, Wijsman EM, Nochlin D, y col. Chromosome 17 and hereditary dementia: linkage studies in three non-Alzheimer familias and kindreds with late onset FAD. Neurology 1997; 48: 949-954. [ Links ]
142. Sumi SM, Bird TD, Nochlin D, Raskind MA. Familial presenile dementia with psychosis associated with cortical neurofibrillary tangles and neurodegeneration of the amygdala. Neurology 1992;42:120-127. [ Links ]
143. Froelich S, Basun H, Forsell C y col. Mapping of a disease locus for familial rapidly progressive frontotemporal dementia to chromosome 17q12-21. Am J Med Genet 1997;74:380-385. [ Links ]
144. Pekkarinen P; Hovatta I; Hakola P y col. Assignment of the locus for PLO-SL, a frontal-lobe dementia with bone cysts, to 19q13. Am J Hum Genet 1998:62:362-372. [ Links ]
145. Ashworth A, Lloyd S, Brown J, Gydesen S, Sorensen SA, Brun A, et al. Molecular genetic characterisation of frontotemporal dementia on chromosome 3. Dement Geriatr Cogn Disord 1999;10[suppl 1]:93-101 [ Links ]
146. Collinge J, Brown J, Hardy J y col. Inherited prion disease with 144 base pair gene insertion II: clinical and pathological features. Brain 1992;2:448-454. [ Links ]
147. Collinge J, Palmer MS, Sidle CL y col. Famillal Pick's disease and dementia in frontal lobe of non-Alzheimer type are not variants of prion disease. J Neurol Neurosurg Psych 1994;57:762-768. [ Links ]
148. Stevens M; van Dujin CM, Knijff P. Apolipoprotein E gene and sporadic frontal lobe dementia. Neurology 1997;48:1526-1529. [ Links ]
149. Gustafson L, Abrahammson M, Grubb A y col. Apolipoprotein-E genotyping in Alzheimer's disease and frontotemporaldementla. Dement Geriatr Cogn Disord 1997;8:240-243. [ Links ]
150. Geschwind D; karrim J; Nelson SF; Miller B. The apolipoprotein E epsilon 4 alíele is not a significant risk factor for frontotemporal dementia. Ann Neurol 1998:44:134-138. [ Links ]
151. Gerst JL, Siedlak SL, Nunomura A, Castellani R, Perry G, Smith MA. Role of oxidative stress in frontotemporal dementia. Dement Geriatr Cogn Disord 1999;10[suppl 1]:85-87 [ Links ]
152. Anderson M, Scott K, Harborne G. Serotonine and depression in frontal lobe dementia [letter], Am J psychiatry 1995:152:645. [ Links ]
153. Swartz JR, Miller BL, Lesser IM, Darby AL. Frontotemporal dementia: treatment response to serotonine selective reuptake inhibitors. J Clin Psychiatry 1997;58:212-216. [ Links ]
154. Sahakian BJ, Coull JJ, Hodges JR. Selective enhancement of executive functions by idaxozan in a patient with dementia of the frontal lobe type. 120-121. [ Links ]
155. Coull JT, Sahakian BJ, Hodges JR: The a2 antagonist idaxozan remediates certain attentional and executive dysfunction in patients with dementia of frontal lobe type. Psychopharmacology 1996:123:239-249. [ Links ]
156. Bettendoríf L, Mastrogiacomo F Wins P, Kish SJ y col. Low thiamine diphosphate levels in brains of patients with frontal lobe degeneration of the non-Alzheimer's type. J Neurochem 1997:69:2005-2010. [ Links ]

