











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
The term sporadic lymphangioleiomyomatosis (S-LAM) is used for patients with LAM which is not associated with the tuberous sclerosis complex (TSC), while TSC-LAM refers to LAM associated with TSC. This rare disorder is part of the perivascular epithelioid cell neoplasms, or PEComas, which are mesenchymal neoplasms made up of histologi cally and immunohistochemically distinctive epithelioid or fusiform cells and are immunoreactive for smooth muscle and melanocyte markers 1.
The diagnosis of LAM is often overlooked and, therefore, there is a significant delay between the onset of the disease and its definitive diagnosis. Any disease showing cysts in the lungs may look like LAM, which is why a lung biopsy has been essential for accurate diagnosis 2.
In the past, biopsies were a common approach to diag nosing LAM; now, there has been a paradigm shift toward less invasive methods that circumvent this procedure, es pecially for S-LAM with no extrapulmonary involvement, as in our case.
Our patient was diagnosed using her medical history, imaging and, mainly, by measuring vascular endothelial growth factor-D (VEGF-D). As far as we know, this is the first time in our country that this factor has been measured for this purpose.
Case presentation
This was a 39-year-old woman from the rural area of the city of Florencia (Caquetá). She was married, with no miscarriages, no children, and no contraceptives; she was a non-smoker and did not use psychoactive substances.
She had a SARS-CoV-2 infection in 2021. She had had difficulty breathing and an occasional cough since the age of 12. She had been hospitalized on several occasions for suspected tuberculosis, but repeated microscopy tests and tuberculin tests were nonreactive, with negative cultures. She was hospitalized several times in Florencia and the city of Neiva. A bronchoscopy with aspirate analysis was not relevant. She reported episodes of pneumonia and one episode of left pneumothorax. She had never had a lung biopsy. She was left with a diagnosis of pulmonary fibrosis of unknown etiology. She was on home oxygen at 2 L/min.
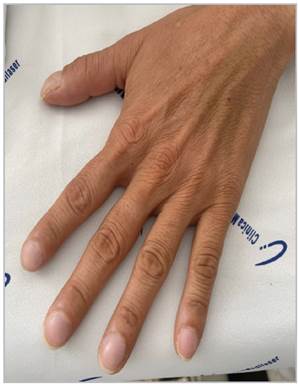
She was admitted with respiratory difficulty and inter costal retractions, low oxygen saturation (88% on 2 L/min of oxygen), oriented and conscious. She had an mMRC 4 dyspnea score, WHO-FC III, with cough and purulent spu tum; on auscultation, she had decreased breath sounds and diffuse crackles; she was afebrile and tachycardic at 124 bpm, with mild nail clubbing (Figure 1).
Laboratory tests were within normal limits, including the following: antinuclear antibodies, extractable nuclear anti-bodies, antineutrophil cytoplasmic antibodies, complement C3 and C4, rheumatoid factor, HIV, CA 125, VDRL, HBsAg, and anti-HCV. Abnormal laboratory tests included the leuko cyte count: 17,800 per microliter with 80.8% nuetrophilia; proBNP: 1,020 pg/mL; and hemoglobin and hematocrit 16.3 g/dL and 54.1%, respectively. The initial arterial blood gases showed: pH 7.31, pCO2 80, pO2 61, HCO3- 40.3, and SaO2 88. The sputum culture grew multisensitive Pseudomonas aeruginosa. Spirometry showed a mixed pattern.
The following tests were not performed: diffusing capacity of the lungs for carbon monoxide (DLCO), serum protein electrophoresis and free light chain quantification, as they are not available in our setting.
Pulmonary hypertension was documented through a suggestive echocardiogram (LV: septal flattening, D sign; RV: dilated chambers, pulmonary artery dilation at 32 mm, PASP 53 mmHg) and proven with a right catheterization (pulmonary artery systolic pressure 52 mmHg, right atrial pressure 12 mmHg, wedge pressure 11 mmHg, generalized hypokinesia and 27% RVEF).
An abdominal-pelvic computed axial tomography (CAT) and head CAT were normal. The chest CAT is shown in Figure 2.
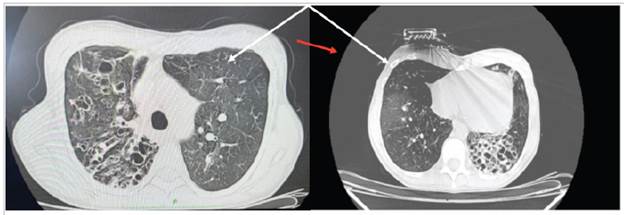
Figure 2 A high-resolution CAT without contrast. Axial plane. Different-sized cystic images. The cysts tend to be round or oval but may be polygonal (red arrow) when there is severe paren chymal involvement. Septal thickening, fibrous scar tracts, bronchiectasis, nodules and ground glass (white arrow) can be seen.
In light of suspected LAM, VEGF-D was ordered at an out-of-town laboratory (Synlab). The value reported was 155.3 pg/mL (verified with the laboratory), which was not significant; therefore, the sample was sent to the Translational Trial Development and Support Laboratory (TTDSL) at Cincinnati Children's Hospital, Ohio (USA), in collabo ration with the LAM Foundation and taking into account the proper sample transport instructions. The new report indicated 1,533 pg/mL (diagnosis > 800 pg/mL). Sirolimus 1 mg/day was added to the usual treatment.
Discussion
The most common signs and symptoms of the disease are caused by pulmonary involvement in S-LAM. Neurocutaneous findings (angiofibromas, shagreen patches, cortical tubercles, seizures and cognitive impairment, among others) are typical of TSC-LAM and are not found in S-LAM 3.
The true incidence and prevalence of S-LAM are unknown, as the available epidemiological data are observational and often include patients with TSC. Consequently, clinical experience and most studies confirm that the sporadic LAM variant is rare and almost exclusively affects women, but many have not yet been diagnosed 4. Previous estimates suggested a rate of one per million in the general population; however, more recent data indicate higher rates, which may reflect advances in recognizing and diagnosing the disease. For example, the LAM Foundation estimated a prevalence of 3-5 per million women, but approximately 10-15% of the patients registered in the foundation report having TSC 5.
The main histopathological abnormality in LAM is prolif eration of atypical smooth muscle-like cells (LAM cells). In the lungs, LAM cells are associated with multiple cysts 2.
Dyspnea on exertion is the most common presenting symptom in LAM. As dyspnea is nonspecific and common, and LAM is rare, women are often labeled with a different diagnosis before suspecting LAM.
There are a variety of pulmonary diseases whose predominant CAT finding is cysts. A cyst is a "round parenchymal transparency or a low attenuation area with a well-defined interface with the normal lung" (6, translated). The differential diagnosis of these diseases is based on the number, size, shape and distribution of these cysts, as well as other associated findings. The characteristic LAM cysts are thin-walled, diffuse, rounded, well-defined, bilateral and with no lobe predominance. The cysts are generally 2-5 mm large but may be larger. They may be round or oval but may become polygonal with severe parenchymal involvement. Small centrilobular nodules have been reported, correspond ing to hyperplastic muscle or pneumocyte hyperplasia. Focal ground-glass opacities may be due to smooth muscle proliferation, and lymphatic obstruction may cause septal thickening 6,7. In the Japanese study, the frequency of pulmonary nodules was higher than in previous reports 8. In our case, we did not find what is typically described; there was more structural disorganization. While the accuracy of a LAM diagnosis based on high-resolution CAT is high for LAM experts 7, we believe that basing the diagnosis solely on the CAT is inadvisable for clinical decision making. A reliable clinical diagnosis of S-LAM is possible when the cystic changes on the CAT are typical for LAM. But we must remember that not all cystic lung diseases are LAM, but all LAM, with time, develops pulmonary cysts. However, we must also be prudent, because not all pulmonary parenchymal transparencies are cysts; there are certain conditions which mimic them (Table 1). Table 2 shows some of the main causes of cystic lung lesions.
Table 1 Pulmonary parenchymal transparencies that mimic cysts. Modified from References 6 and 7.
| Definitions | Characteristics | Examples |
| Bulla | Round, focal, thin-walled (<1 mm); generally several centimeters in diameter, may fill the hemithorax, may be associated with emphysema. | Emphysema. |
| Bronchiectasis | Dilated, thick-walled bronchi may appear cystic, but they communicate with the airways; may be grouped. | Cystic fibrosis, primary ciliary dysfunction, common variable immuno deficiency, postinfectious. |
| Cavitation | The thick walls (> 4 mm) may be partially filled with fluid, debris and eumycetoma. May be within a mass, nodule or area of consolidation. | Necrotizing bacterial pneumonia, mycobacterial or fungal infections, neoplasms (e.g., bronchogenic carcinoma, if single; metastatic cancer, if multiple), granulomatosis with polyangiitis or other vasculitides. |
| Cyst | Round or irregular, varying in size, with thin walls (<2 mm thick), may be associated with nodules. | Birt-Hogg-Dubé syndrome, lymphangioleiomyomatosis, lymphocytic interstitial pneumonia, pulmonary Langerhans cell histiocytosis. |
| Emphysema | Round or polygonal lucencies with no walls; variable size. Paraseptal emphysema is delineated by interlobular pleura and septa: centrilobular emphysema has a predilection for the upper lobe. | Alpha-1 antitrypsin deficiency, pulmonary lesion due to smoking ciga rettes or cooking with wood (biomass). |
| Honeycomb | Clusters of round, 3-10 mm diameter lucencies, with a 1 to 3 mm wall thickness; may have several levels, generally in a subpleural location, associated with traction bronchiectasis and septal thickening. | Asbestosis, idiopathic pulmonary fibrosis, interstitial pneumonia typical of systemic rheumatic disease, chronic hypersensitivity pneumonitis. |
| Pneumatocele | Often postinfectious, may be single or multiple, typically transient. | Postinfectious (e.g,, Staphylococcus aureas pneumonia, Pneumocystis jirovecii pneumonia, coccidioidomycosis), postraumatic, hydrocarbon inhalation. |
Table 2 Causes of cystic pulmonary lesions. Modified from Reference 7.
| Congenital | Immune |
|---|---|
| Cystic adenomatoid malformation | Wegener's granulomatosis |
| Pulmonary cyst | Lymphocytic interstitial pneumonia |
| Birt-Hogg-Dubé syndrome | Follicular bronchiolitis |
| Infectious | Autoimmune disease |
| Staphylococcus aureus pneumonia | Primary Sjogren syndrome |
| Melioidosis | Neoplasms |
| Histoplasmosis | Squamous cell carcinoma |
| Idiopathic | Invasive mucinous adenocarcinoma |
| Langerhans cell histiocytosis | Cystic metastases |
| Amyloidosis | Angiosarcoma |
| Light chain deposition disease | |
| Others | |
| Centrilobular emphysema | |
| Lymphangioleiomyomatosis | |
| Pulmonary sequestration | |
Given the serum VEGF-D biomarker' s good performance and taking the recommended diagnostic threshold (>800 pg/mL) for its sensitivity and specificity of 73-100%, respectively, rules out other pulmonary cystic lung diseases which are commonly considered in the differential diagnosis, as they do not significantly elevate the VEGF-D, and this avoids 70% of biopsies 9. Studies so far have not found false positives, but a negative result does not rule out the diagnosis 9,10.
The following are notable in this case:
The chest CAT does not show the typical images found in these patients, but sometimes what is typical is not what is usual in the real world. We believe that we should not rule out the disease if we do not have the so-called "typical" images, especially if we do not have a radi ologist who is an expert in pulmonary cysts or LAM. When in doubt, a biomarker should be ordered before performing a biopsy. In our case, we had atypical images with typical VEGF-D levels from a LAM sensitivity and specificity perspective 10. In patients with S-LAM, like our patient, and with no other manifestation like renal angiomyolipomas, chylous fluid accumulation or TSC, a diagnosis cannot be made using high-resolution CAT, because the precision is estimated to be < 80% with expert radiologists 11.
The finding of Group 3 pulmonary hypertension. It has been described as precapillary and is generally an uncommon finding in people with LAM, < 7%, and, when found, is mild 12, unlike our case, which is why the di agnosis was questioned before obtaining the VEGF-D. A lung biopsy was proposed, which the patient refused after learning the benefits and risks of the procedure. Pulmo nary involvement has been reported to be more severe in S-LAM 12. Our patient's pulmonary hypertension was likely due to hypoxic pulmonary vasoconstriction with pulmonary vascular bed remodeling, along with vascular involvement due to inflammatory parenchymal fibrosis caused by repetitive infections, together with bronchial damage due to LAM cell and lymphatic proliferation, in which chronic inflammation tends to complicate the disease. Keeping in mind that LAM patients show pro gressive cystic destruction of the lung parenchyma.
Nail clubbing is not generally one of the physical findings in LAM 13; however, it may have been found in our patient due to chronic cardiopulmonary involvement.
A mixed pattern on spirometry. This is not usual, either; it only occurs in less than one-fourth of cases. Most are obstructive, followed by normal 13.
Two different VEGF-D results. In the first test, we do not know if correct sample packaging and processing pro cedures were followed. This is why another report was sought from a more sophisticated laboratory specialized in these areas, given the persistent clinical suspicion.
Recurrent pneumothorax, according to different authors, is characteristic of LAM, with an incidence of 61 to 81% 13. This was not a finding in our case, either; according to her history, she had only had one episode of pneumothorax.
In conclusion, we have a patient with S-LAM diagnosed by high VEGF-D levels, but who does not have the usual clinical and tomographic characteristics reported in the literature. Therefore, the disease should not be ruled out in patients who do not have the described characteristics, until the diagnostic tools have been exhausted, especially the serum biomarker. In appropriate clinical situations, the VEGF-D test is a significant advance in diagnosis and current effective treatment, and may make a lung biopsy unnecessary for diagnosing LAM. While there is no cure, living with the disease is very different today than in the past; where there was anxiety and fear there is now hope - it is no longer a death sentence.
What exists today is an unmet need for additional bio-markers of disease activity and/or response to treatment. These biomarkers would allow personalized and precision clinical care in the future.
Ethical aspects
For this article, informed consent was obtained from the patient according to the ethical principles contained in the Declaration of Helsinki. The article was also approved by the Ethics Committee at Clínica Mediláser, Florencia branch, Florencia, Colombia.

