











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkINTRODUCTION
Dental enamel is an acellular, avascular, and non-inert structure consisting of a mineral phase and an organic form composed of structure proteins, lipids, phospholipids, enzymes, and water.1 During enamel formation, there can be alterations of the enamel structure responsible for aesthetic and functional problems. One of these alterations is amelogenesis imperfecta (AI), a condition of genetic origin that impacts the structure of tooth enamel and can appear as a syndromic alteration or isolated. When the enamel alteration affects a group of teeth from both dentitions and there are systemic clinical manifestations, the etiological origin is likely due to genetic alterations.2
AI has an autosomal dominant, autosomal recessive, X- and Y-linked inheritance or sporadic pattern. According to the scarce epidemiological studies available, the prevalence of AI can vary from 1/700 to 1/14.000 in countries like Switzerland and the United States respectively.3 Alterations in the dental structure are usually widespread and affect all or most teeth. There are three forms of AI: hypoplastic, hypocalcified, and hypomaturation. Various AI classifications have been proposed, but the Witkop4 classification is still widely accepted as it includes aspects like phenotype, radiographic appearance, and mode of transmission.
The clinical manifestations of AI include loss of enamel translucency, tooth hypersensitivity, yellow, gray or brown pigmentation, and early attrition at the incisal level and at the cusps of molars. Dental eruption and gingival alterations are common, as well as taurodontism; also, teeth with AI are more likely to be affected by carious processes as a result of biofilm retention.5,6 Given the importance of AI as an alteration of tooth enamel affecting patients of different age groups, our goal is to describe the genes and mutations involved in syndromic AI, the proteins encoded by these genes, and their functions according to current scientific evidence.
METHODS
An electronic literature search was conducted in the Medline (PubMed), EBSCOhost and Scopus (Science Direct) databases using the following keywords: amelogenesis imperfecta, genes AND/OR amelogenesis imperfecta, and syndrome AND/OR amelogenesis imperfecta. Full articles in English, accessible in PDF and produced from 2000 until December 2017 were obtained as inclusion criteria. No age or sex restrictions were used. Dissertations, newspapers, conferences, news, commentaries, and editorials were excluded.
The articles used in this literature review were collected and stored using the Zotero software, which also allowed to eliminate duplicate bibliographic references. The articles were independently evaluated by two reviewers according to the above criteria. Finally, a screening was conducted applying the criteria to the articles found in the databases, and 1,573 articles were preselected; of these, 40 met the inclusion criteria and were therefore analyzed and discussed (Table 1).
Table 1 Summary of the approach used for the electronic literature search
| Keywords | Medline (PubMed) | EBSCOhost | Science Direct (Elsevier) |
|---|---|---|---|
| Amelogenesis imperfecta | 262 | 400 | 219 |
| Gene and/or Amelogenesis imperfecta | 138 | 194 | 142 |
| Syndrome and/or Amelogenesis imperfecta | 53 | 92 | 73 |
Source: Simancas V, Natera A, Acosta MG
RESULTS
The results showed that mutations in 12 genes are responsible for syndromic AI. Below is a description of each gene.
Genes involved in syndromic amelogenesis imperfecta
1. DLX3
The DLX3 gene (OMIM 600525, distal-less homeobox 3) has 3 exons and is located on chromosome 17, position 17q21.33. This gene encodes for a protein that is key in regulating the expression of osteocalcin during embryogenesis, along with Dlx3, Dlx5, Msx2 and Runx2. DLX3 has shown to be an enhancer in the expression of amelogenin and enameline. DLX3 expresses in bones and dental tissues, particularly in odontoblasts and ameloblasts during the secretion stage.7
The DLX3 gene mutation produces a hypomaturation-hypoplastic AI with broad pulp chambers and taurodontism. Indeed, this gene is reported to be the cause of the tricho-dento-osseous syndrome (TDO). TDO belongs to the ectodermal dysplasia group, characterized by curly hair from birth, enamel hypoplasia with discoloration and taurodontism in molars, overall increase in bone mineral density, and increased thickness in cortical skull bones.8,9
Mutations in the COL17A1, LAMA3, LAMB3 genes are responsible for junctional epidermolysis bullosa (JEB) and AI. JEB is a form of hereditary epidermolysis bullosa characterized by the formation of blistery lesions between dermis and epidermis, and by the healing of lesions with atrophy and/or formation of granulation tissue.10-12
2. COL17A1
The COL17A1 gene (OMIM 113811, collagen, type XVII, alpha-1) is located in chromosome 10, position 10q24,3-q25.1. It has 56 exons and encodes for type XVII collagen. Nakamura et al13 reported two patients with JEB and a dental phenotype consistent with hypoplastic AI. The first patient had a deletion at position c.1.285 in exon 15 which resulted in the presence of a premature termination codon. This same patient also had a transition of cytosine to thymine at position c.4.264 in exon 53 that caused the conversion of codon CAG into a premature termination codon in amino acid 1.387. In the second patient, the COL17A1 gene mutation occurred at position c.4.335 in exon 53, a deletion that led to the presence of a premature termination codon.
In a report describing different forms of JEB in the Netherlands caused by mutations in the COL17A1 gene, Pasmooij et al14 identified five new deletions: 1.284delA, 1.365delC, 3.236delT, 3.600-3601delCT and 4.425delT.
Phenotypically, patients had hypoplastic AI and localized or widespread blisters on hands, feet, and face.
3. LAMA3
The LAMA3 gene (OMIM 600805, laminin, alpha-3) consists of 75 exons and is located on chromosome 18, position 18q11.2. This gene encodes for laminin alpha-3, which is also a subunit of laminin 5. Mutations in this gene are related to non-syndromic and syndromic AI. In syndromic AI, mutations of the LAMA3 gene are identified by a hypoplastic AI along with JEB. Gostyńska et al15 reported a nonsense mutation of the LAMA3 gene (c.2.377C4T p. Arg793Ter) in a 2-year-old boy who since the age of 6 months developed blisters on hands, legs and ears, along with a hypoplastic AI. Yuen et al10 reported dental enamel defects related to hypoplastic AI in a mutation-carrying family in the LAMA3 gene. The report showed the c.488delG mutation in exon 5, which produced a reading frame change and consequently a premature termination codon (p. G163DfsX30). The mutation c.4.484C>T in exon 33 of the LAMA3 gene- which induces a replacement of alanine to valine at position 1.495 p. A1.495V-was also reported.
4. LAMB3
The LAMB3 gene (OMIM 150310, laminin, beta-3) is composed of 23 exons and is located on chromosome 1, position 1q32.2. This gene encodes a protein known as laminin beta-3, a subunit of laminin 5. It acts at the transmembrane (in the basal membrane) and participates in cell growth and adhesion.16 It has been established that the mutations of the LAMB3 gene are responsible for syndromic and non-syndromic AI. Within the syndromic AI, changes in the LAMB3 gene are also responsible for the presence of AI and JEB. Clinically, patients have internal skin blisters caused by an alteration in the formation of hemidesmosomes between the epidermis and basal lamina, along with enamel defects of varying severity.12
5. FAM20A
The FAM20A gene (OMIM 611062, family with sequence similarity 20, member A) has 11 exons and is located on chromosome 17, position 17q24.2. FAM20A is a gene belonging to the family with similarity in sequence 20, called FAM20, as well as FAM20B and FAM20C.17 Recently, Cui et al18,19 demonstrated that FAM20A enhances the activity of the protein kinase FAM20C which promotes the phosphorylation of proteins in the enamel matrix in vitro. According to Ohyama et al,20FAM20A is a pseudo-kinase that forms a functional complex with FAM20C, and thanks to this functional complex, FAM20C increases its phosphorylation capacity. As a result, mutations of the FAM20A gene contribute to deregulation of the cellular microenvironment of calcium and/or phosphate by favoring an alteration of the regulatory processes of mineralization.21
The autosomal recessive mutation in the FAM20A gene is responsible for the Amelogenesis Imperfecta-Nephrocalcinosis Syndrome, also known as Enamel- Renal Syndrome (ERS - OMIM 204690) which associates a hypoplastic AI with nephrocalcinosis. This syndrome affects both deciduous and permanent teeth. In addition to hypoplastic AI, this syndrome has other clinical conditions, including gingival hyperplasia, alterations in teeth eruption, relative microdontia, spaced-out teeth, intrapulp calcification, dilation of impacted teeth roots, and ectopic calcification of dental and gum follicles, coupled with nephrocalcinosis.22
6. TP63
The TP63 gene (OMIM 603273, tumor protein P63) has 14 exons and is located on chromosome 3, position 3q28. It is responsible for the coding of an essential transcription factor that initiates ectodermal development, as well as limb and tooth development. This transcription factor is secreted by epithelial cells and is involved in the transfer of signals to the ectomesenchymal cells.23
TP63 mutations have been linked to the ectrodactyly-ectodermal dysplasia clefting syndrome (EEC syndrome), a genetic developmental disorder characterized by ectrodactyly, ectodermal dysplasia, and orofacial clefts (cleft lip/palate).23,24 Kantaputra et al25 reported on a family with a nonsense heterozygous mutation in intron 4 of the TP63 gene where, in addition to ectodermal dysplasia and ectrodactyly, one of the patients showed supernumerary premolars, incisor fusion and hypoplastic, hypocalcified and hypomaturation AI.
7. CNNM4
The CNNM4 gene (OMIM 607805, cyclin protein M4) is composed of 7 exons and is located on chromosome 2, position 2q11.2. This gene encodes a protein of 775 amino acids involved in the transport of metal ions, most likely magnesium (Mg), essential for the proper functioning of photoreceptors in the retina. CNNM4 is expressed in the ameloblasts and odontoblasts, and therefore its presence is fundamental in the formation of tooth-enamel crystals.26,27
Autosomal recessive mutations of the CNNM4 gene result in the Jalili Syndrome, characterized by a combination of hypoplastic and/or hypocalcified AI, along with cone-rod dystrophy. The enamel appears cracked and rough with a yellow coloration. An anterior open bite and taurodontism of the molars are also common. The cone-rod dystrophy, on the other hand, causes central vision loss, color vision defects, and photophobia long before the age of 10.28,29
8. ROGDI
The ROGDI gene (OMIM 614574, ROGDI homologous of Drosophila OG) has 7 exons and is located on chromosome 16, position 16p13.3. It encodes for a leucine zipper domain protein. The role of the ROGDI protein is still unknown, but it is known to interact with MAP kinase (mitogen-activating protein) in the cell cycle. Also, ROGDI interacts with DISC1, a protein involved in cytoskeleton stability, neuronal migration, intracellular transport and cell division.30,31
As a result of an autosomal recessive mutation, the ROGDI gene is responsible for amelo-cerebro-hypohidrotic syndrome or Kohlschütter-Tönz Syndrome, a degenerative disorder characterized by AI, convulsions, and yellow teeth due to enamel defects. Its clinical manifestations may be limited to cognitive problems, speech delay and learning problems, or may be associated with psychomotor delay, spasticity and epilepsy. In the mouth, it shows a hypocalcified-hypoplastic AI along with a very thin enamel.32,33
9. LTBP3
The LTBP3 gene (OMIM 602090 latent transforming growth factor-beta-binding protein 3) has 28 exons; it is located on chromosome 11, position 11q13.1 and encodes for a protein of the TGF-beta signaling pathway, involved in the production and degradation of the extracellular matrix. The autosomal recessive mutation of the LTBP3 gene produces brachyolmia-amelogenesis imperfecta. This pathology is part of a heterogeneous group of skeletal dysplasia characterized by a short stature and a short trunk. A hypoplastic AI with taurodontism and wide pulp chambers, delayed dental eruption, and oligodontia are part of the phenotypic characteristics of these patients.34-36
10. FAM20C
The FAM20C gene (OMIM 611061, family with sequence similarity 20, member C) consists of 10 exons. Located on chromosome 7, position 7p22.3, FAM20C is a gene belonging to the family with sequence similarity 20, called FAM20, which encodes for a casein kinase of the golgi apparatus responsible for phosphorylating enamel proteins (amelogenins, enamelins and ameloblastins), osteopontin, and a large amount of proteins involved in various physiological processes.37-39
The recessive FAM20C mutation leads to Raine Syndrome, a condition characterized by a hypophosphatemia disorder, generalized osteosclerosis, periosteal ossification, facial dimorphism, and intracerebral abnormalities. Gingival hyperplasia, bifid uvula, cleft palate, and hypoplastic AI are part of the oral phenotype. It was initially considered a lethal pathology at an early age, but patient survival into adulthood has been reported.40
11. CLDN16
The CLDN16 gene (OMIM 603959, claudin 16) has 5 exons and is located on chromosome 3, position 3q28. It encodes for a protein that plays a central role in the reabsorption of divalent cations in the tight junctions of renal epithelial cells of the ascending limb of Henle, where it forms selective cation pores that facilitate the reabsorption of Ca2+ and Mg2+.41,42
The CLDN16 mutation is responsible for familial primary hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHN), a form of familial primary hypomagnesaemia characterized by recurrent urinary tract infections, nephrolithiasis, bilateral nephrocalcosis, renal loss of Ca2+ and Mg2+, hypercalciuria and kidney failure. Recently, Bardet et al43) demonstrated the expression of CLDN16 in dental germ, showing that mutations of this gene are responsible for AI in the 5 FHHNC studied patients. The dental examination showed that all patients had enamel defects, including a decrease in the amount of enamel; in fact, four patients had hypoplastic AI and one of them had hypomaturation AI.
12. CLDN19
The CLDN19 gene (OMIM 610036, claudin 19) has5exonsandis locatedonchromosome 1, position 1q34.2. It is responsible for encoding transmembrane proteins found in tight junctions of renal epithelial cells just like CLDN16. These junctions form barriers that control the passage of ions and molecules through epithelial sheets and the movement of proteins and lipids between the apical and basolateral domains of epithelial cells.44
Like mutations in the CLDN16 gene, the CLDN19 mutation is responsible for familial primary hypomagnesaemia with hypercalciuria and nephrocalcinosis. In 2017, Yamaguti et al45 reported six families unrelated to CLDN19 mutations. Clinically, a hypoplastic AI with nearly complete absence of enamel, crown dysmorphology, dark brown teeth, and enamel fractures were described.
A summarizing table displays information regarding the known genes involved in AI (Table 2), while Figure 3 shows the function of proteins encoded by those genes according to anatomical compartment during tooth enamel formation.
Table 2 Genes involved in syndromic amelogenesis imperfecta
| Gene (OMIM) | Position | Number of exons Transmission | Encoded protein | AI type/associated syndrom |
|---|---|---|---|---|
| DLX3 (distal-less homeobox 3) OMIM 600525 | 17q21.33 | 3-AD | Transcription factor | Hypoplastic and Hypomaturation / trichodento-osseous syndrome (TDO) |
| COL17A1 (collagen, type XVII, alpha-1) OMIM 113811 | 10q24,3-q25.1 | 56-AD | Collagen, type XVII, alpha1 | Hypoplastic / Junctional Epidermolysis Bullosa (JEB) |
| LAMA3 (laminin, alpha-3) OMIM 600805 | 18q11.2 | 75 -AR | Laminin, alpha-3 | Hypoplastic / Junctional Epidermolysis Bullosa (JEB) |
| LAMB3 (laminin, beta-3) OMIM 150310 | 1q32.2 | 23-AR | Laminin, beta-3 | Hypoplastic / Junctional Epidermolysis Bullosa (JEB) |
| FAM20A (family with sequence similarity 20, member A) OMIM 611062 | 17q24.2 | 11-AR | Secreted pseudokinase | Hypoplastic / Amelogenesis imperfect Nephrocalcinosis Syndrome or Enamel-Rena Syndrome (ERS) |
| TP63 (tumor protein P63) OMIM 603273 | 3q28 | 14-AD | Transcription factor | Hypoplastic, hypomaturation and hypocalcified / Ectrodactyly-Ectodermal Dysplasia Clefting Syndrome (ECC) |
| CNNM4 (cyclin protein M4) OMIM 607805 | 2q11.2 | 7-AR | Metal transporter | Hypoplastic and hypocalcified / Jalili Syndrome |
| ROGDI (ROGDI homologous of Drosophila OG) OMIM 614574 | 16p13.3 | 7-AR | Unknown functional role | Hypocalcified and hypomaturation / Amelo-cerebro-hypohidrotic syndrome or Kohlschütter-Tönz Syndrome |
| LTBP3 (Latent-transforming growth factor beta-binding protein 3) OMIM 602090 | 11q13.1 | 28-AR | Production and degradation of extracellular matrix | Hypoplastic with taurodontism / Brachyolmia |
| FAM20C (family with sequence similarity 20, member C) OMIM 611061 | 7p22.3 | 10-AR | Protein kinase | Hypoplastic / Raine Syndrome |
| CLDN16 (Claudin 16) OMIM 603959 | 3q28 | 5-AR | Tight joints of renal epithelial cells | Hypoplastic and hypomature / Familial primary hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHN) |
| CLDN19 (claudin 19) OMIM 610036 | 1q34.2 | 5-AR | Tight joints of renal epithelial cells | Hypoplastic / Familial primary hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHN) |
AD: autosomal dominant; AR: autosomal recessive
Source: Simancas V, Natera A, Acosta MG
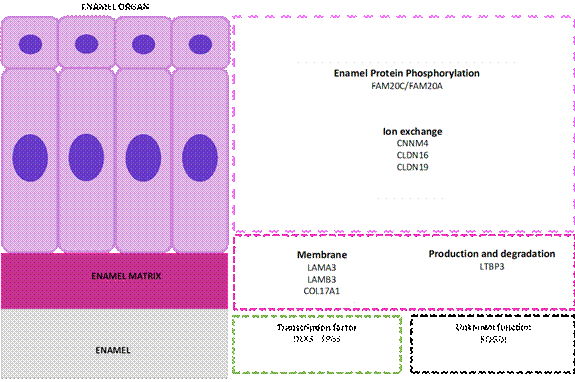
Source: Simancas V, Natera A, Acosta MG.
Figure 3 Function of the proteins encoded by genes involved in amelogenesis imperfecta. The enamel organ with ameloblastic cell area (purple), enamel matrix (violet), and enamel (grey). In the lower area, the transcription factors (green box) and genes responsible for amelogenesis imperfecta with no currently known function at the dental level (black box).
CONCLUSIONS
Observation of the bucco-dental and systemic phenotype remains a good indicator in human genetics research. This literature review showed that 12 genes are responsible for syndromic AI, including genes DLX3, COL17A1, LAMA3, LAMB3, FAM20A, TP63, CNNM4, ROGDI, LTBP3, FAM20C, CLDN16, CLDN19. These genes participate in the coding of proteins involved in phosphorylation, ion exchange, production and degradation of constituent elements of the mineral and organic phases of tooth enamel.
In the presence of a phenotype compatible with AI and systemic alterations, a probable genetic origin should be confirmed. For this reason, comprehensive clinical, radiographic and genetic examinations of parents and siblings are recommended, as it will help establish a family tree to specify the mode of transmission. The clinical and radiographic evaluation of kidneys, lungs, vertebrae, and vascular system will help diagnose a syndromic or non-syndromic AI.

