











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
La poliarteritis nudosa (PAN) fue descrita por primera vez por Kussmaul y Maier en 1866[1]. Se define como una vasculitis necrosante de vasos de tamaño mediano/pequeño, sin vasculitis en arteriolas, vénulas y capilares, no asociada con anticuerpos citoplásmicos antineutrófilo (ANCA)[2]. Es la tercera vasculitis más común en niños, después de la vasculitis IgA/púrpura de Henoch-Schonlein y la enfermedad de Kawasaki[3]. En adultos, la incidencia de PAN es de alrededor de 0,0-1,6 casos, mientras que su prevalencia es de aproximadamente 31 casos por millón[4]; es menos frecuente en la infancia, aunque los datos epidemiológicos son escasos en niños[5]. Presenta un pico de edad de inicio en los niños de 9-10 años y ambos géneros son igualmente afectados[6,7]. La PAN del adulto con frecuencia se asocia a infecciones por virus hepatotropos, mientras que en la edad pediátrica el estreptococo y algunos virus han sido postulados como posibles desencadenantes, sin evidencia concluyente[8,9].
Hay 2 presentaciones de la PAN en la infancia. La PAN cutánea, autolimitada en la mayoría de los casos, que puede manifestarse con livedo reticularis, púrpura, hemorragias en astilla, isquemia digital, nódulos dolorosos, artritis, y miositis; en general, los órganos internos no se ven afectados[8]. La PAN sistémica tiene un espectro de presentación más grave y, además de las manifestaciones de la PAN cutánea, los pacientes pueden presentar dolor abdominal y angina intestinal, que en algunos casos progresan a hemorragia intestinal, infarto y perforación intestinal[3,8]. La afectación de los vasos renales conduce a hipertensión e insuficiencia renal, raramente también ocurren el dolor y el infarto testicular; a nivel neurológico el sistema nervioso periférico suele comprometerse en forma de mononeuritis múltiple, mientras que el sistema nervioso central suele no tener afectación. También se ha descrito compromiso ocular, mientras que a nivel pulmonar, en general, no hay compromiso, lo que ayuda a diferenciarla de otros tipos de vasculitis.
La mortalidad por PAN es baja (alrededor de 4%) y el curso puede ser de remisión completa o recaída-remisión[3,6-8].
Las pruebas de laboratorio son inespecíficas; durante la actividad de la enfermedad se pueden encontrar leucocitosis, trombocitosis y elevación de reactantes de fase aguda. Los autoanticuerpos generalmente son negativos, incluidos los ANCA[10].
La PAN infantil se clasifica de acuerdo con los criterios EULAR/PRINTO/PRES revisados en 2010[11]; cabe resaltar que estos son criterios de clasificación y no diagnósticos; en estos, la histopatología típica de vasculitis necrosante o las anomalías angiográficas son esenciales en el diagnóstico, con una sensibilidad del 84 y el 81%, respectivamente, y una especificidad de 99% para las 2[11]. Los hallazgos clínicos incluyen compromiso en la piel (livedo reticularis, nódulos subcutáneos, infartos superficiales y profundos, cambios isquémicos menores, necrosis/gangrena), dolor o sensibilidad muscular, hipertensión (presión arterial sistólica/diastólica > percentil 95), neuropatía periférica sensorial o mononeuritis y compromiso renal dado por proteinuria, hematuria y alteración en la tasa de filtración glomerular[11].
Aunque la angiografía convencional sigue siendo el «estándar de oro» y tiene la sensibilidad más alta para detectar estenosis en la vasculatura más pequeña y microaneurismas, es una técnica invasiva con una significativa exposición a altas dosis de radiación. Es por eso por lo que la angiografía por resonancia magnética y la angiografía tomográfica computarizada se utilizan cada vez más en niños[11,12].
La mayoría de los tratamientos para niños se extrapolan de ensayos en adultos[13]. Se utiliza tratamiento de inducción con glucocorticoides y ciclofosfamida (CYC) y de mantenimiento con azatioprina (AZA) por vía oral, metotrexato o leflunomida. El uso de agentes biológicos en cohortes retrospectivas ha mostrado ser eficaz y seguro en niños con vasculitis sisté-mica primaria. No obstante, aún se requieren protocolos de tratamiento y ensayos clínicos[14,15].
Caso clínico
Paciente femenina de 4 años, previamente sana, sin antecedentes patológicos relevantes, con cuadro inicial de fiebre y odinofagia y diagnóstico de amigdalitis bacteriana (no se confirma infección por Streptococcus); recibió manejo antibiótico convencional de manera ambulatoria.
Durante los 6 meses siguientes consulta en múltiples ocasiones a urgencias por cuadros de fiebre de 3-4 días de duración, de 38-39°C, asociado en su mayoría a dolor abdominal posprandial y dolor en miembros inferiores, en ocasiones incapacitante. Los estudios de primera línea realizados son normales, ocasionalmente hay en el hemograma leucocitosis leve y trombocitosis, elevación de reactantes de fase aguda, principalmente VSG, sin compromiso franco del estado clínico, por lo que recibía manejo ambulatorio.
Luego de 8 meses del cuadro inicial presentó fiebre prolongada de 22 días de duración, acompañada de dolor abdominal posprandial y agudización de mialgias intensas, que impedían de manera intermitente la deambulación, por lo que requirió hospitalización. En el examen físico se encontraron fenómeno de Raynaud de reciente aparición y livedo reticularis como únicos hallazgos.
Se realizó un estudio de extensión completo, descartando causas infecciosas comunes y no comunes; malignidad con estudio de médula ósea, que fue negativo para células blásticas, tumorales y hemofagocitos, biopsia de piel compatible con livedo reticularis y estudio inmunológico con anticuerpos negativos y complemento normal. Luego de 26 días de estudios dentro de la normalidad, dada la resolución de la fiebre y el adecuado estado clínico de la paciente se continuaron estudios de manera ambulatoria que también fueron negativos.
Luego de un año de seguimiento irregular con episodios de fiebre de 1 a 2 por mes, presentó disminución de agudeza visual en el ojo derecho, con hallazgo de atrofia del nervio óptico por Oftalmología. Los estudios para autoinmunidad fueron nuevamente negativos.
Dos meses después, asiste a urgencias con cuadro de fiebre, dolor abdominal y cefalea con urgencia hipertensiva, que requiere manejo en la Unidad de Cuidado Intensivo (UCI) por varios días por hipertensión arterial (HTA) de difícil manejo (estudio para causas secundarias de HTA normales). Paraclínicos con leucocitosis, trombocitosis y elevación importante de reactantes de fase aguda (VSG y PCR). Durante la estancia en la UCI presenta angina mesentérica (no fue posible toma de angiografía por la condición clínica del paciente). El análisis de las manifestaciones clínicas y la severa respuesta inflamatoria sistémica permitieron considerar como opción diagnóstica una vasculitis sistémica tipo PAN. Se inició manejo con glucocorticoide en pulsos (número de 3) y se continuó a 1 mg/k/día asociado al manejo de HTA con 2 medicamentos dado el difícil control de cifras tensionales.
Durante el seguimiento, la paciente recibe tratamiento con esteroides a dosis de 1 mg/k/día, presentando una actividad leve persistente, con reactivaciones; en una de ellas, compromiso renal, por lo que se decidió inicio de CYC por vía intravenosa; recibió 6 dosis mensuales y prednisolona a dosis altas (2 mg/k/día), con lo cual se logra llevar a remisión. El tratamiento de mantenimiento tuvo dificultades debido a la adherencia, la intolerancia a metotrexato y falla a AZA, por lo cual es llevada a junta de Reumatología Pediátrica y se decide iniciar CYC por vía oral (2 mg/kg/día), logrando el control de la enfermedad.
Luego de 5 años desde el inicio de la enfermedad (9 años de edad), el seguimiento es irregular por problemas psicosociales (condiciones económicas, trámites administrativos, mala adherencia, etc.). Recibe CYC por vía oral de forma discontinua y dosis variables de prednisolona, sin lograr descenso efectivo. En este momento la paciente inicia cuadros de hematuria y síntomas irritativos urinarios bajos. El estudio para hematuria indica etiología no glomerular y compromiso bajo; la ecografía renal es normal. Dado el antecedente de uso prolongado de CYC por vía oral (aunque con dudas sobre adherencia), a una dosis acumulada de 100 g en 5 años, y el riesgo de efectos secundarios relacionados con la CYC (cistitis hemorrágica [CH]), se decide suspender la CYC, reiniciar AZA y evaluar la respuesta.
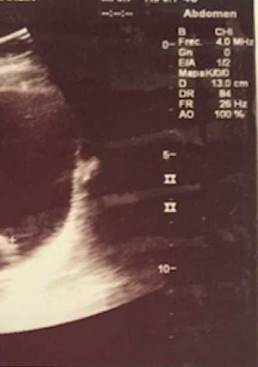
A pesar de la suspensión de CYC, luego de 8 meses, continúan los episodios recurrentes de hematuria, uno de ellos severo, que compromete el estado hemodinámico de la paciente, con disminución de hemoglobina hasta 7 g/dl y presencia de un gran coágulo intravesical (fig. 1). Se ampliaron los estudios por Nefrología y Urología pediátrica para otras causas de hematuria persistente, entre ellos polioma-virus, adenovirus, micobacterias, nefritis intersticial, cistitis eosinofílica, etc., los cuales fueron negativos. La uro-TAC documentó engrosamiento leve y difuso de las paredes de la vejiga; teniendo el estudio negativo para infección y persistencia de la sintomatología, se decidió realizar una cistoscopia, en la que se observó una mucosa hiperémica con vasculitis generalizada de la mucosa vesical.
La histopatología vesical mostró un patrón predominante de daño vasculítico (figs. 2 y 3), con necrosis de las paredes vasculares, con edema endotelial asociado y marcada respuesta inflamatoria secundaria, leucocitoclástica, hemorragia y necrosis (fig. 4).
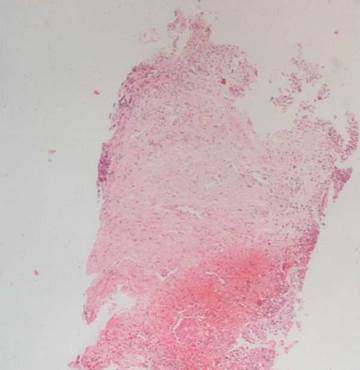
Figura 2 Panorámica vesical H&E 10 x. Se observa pared vesical con mucosa despulida, hay hemorragia extensa y no se observan claramente las paredes de los vasos sanguíneos.
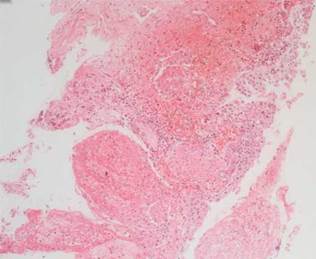
Figura 3 Ampliación vesical H&E 40 x. Proceso vasculítico instaurado; en los vasos sanguíneos vesicales se observa necrosis, fibrina e infiltrado inflamatorio vascular y perivascular.
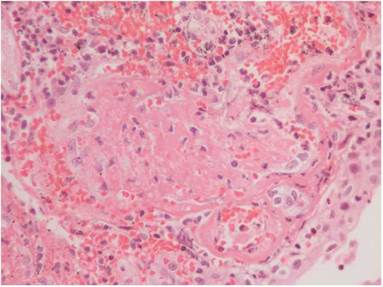
Figura 4 Vasculitis y necrosis H&E 10 x. Pared vesical, con vasos sanguíneos que muestran proceso de necrosis parcial de la pared, hemorragia, inflamación en pared vascular, pérdida del urotelio de revestimiento y extensa hemorragia de la pared.
El compromiso inusual y severo en paciente con refractariedad a otros tratamientos y un tiempo prolongado de esteroides replanteó el manejo terapéutico; nuevamente, en junta de Reumatología, apoyados por la literatura (aunque escasa), se inició manejo con rituximab a dosis de 750mg/m2 en las semanas 0 y 2, asociado a CYC 500 mg por vía intravenosa y glucocorticoide, con lo cual se logró el control de la complicación y se alcanzó la remisión.
Actualmente, a sus 16 años, la evolución de la paciente ha sido favorable, ha tolerado la terapia de mantenimiento con AZA, sin nuevos episodios de hematuria, con mejor adherencia, entendimiento y control de su enfermedad, y permitió la disminución progresiva de la dosis de esteroide, hasta la suspensión en la actualidad.
Discusión
La PAN es considerada un diagnóstico de exclusión, caracterizado por presentar compromiso multisistémico, con síntomas comunes a otras afecciones (infección, malignidad, etc.)[16], requiere un alto índice de sospecha y representa un verdadero desafío diagnóstico para los clínicos.
Las características de la presentación difieren de acuerdo con los órganos involucrados[7], aunque cualquier sistema puede estar involucrado. Las características clínicas en la presentación de una cohorte de PAN sistémica en pediatría en 69 niños fueron las siguientes: fiebre (87%), mialgia/sensibilidad muscular (83%), piel (88%), renal (19%), gastrointestinal grave (10%) y afectación neurológica (10%)[6]. La hipertensión se describe entre un 43 a un 90% en las diferentes cohortes de pacientes pediátricos con PAN y en general es de difícil manejo[6,7,17,18].
En Colombia se reportó en 2017 una cohorte de 22 niños[19] en la que las manifestaciones más frecuentes al inicio fueron compromiso articular en el 72% de los pacientes, seguido de síntomas constitucionales fiebre y mialgias (68%) y manifestaciones cutáneas; el compromiso orgánico estuvo liderado por el renal y el gastrointestinal, este último con manifestaciones severas, como la angina intestinal.
Los nódulos cutáneos son la manifestación cutánea más común en pacientes con PAN juvenil; se presentan como nódulos eritematosos en las extremidades inferiores, miden de 1 a 5 cm de diámetro y pueden unirse para formar grandes áreas de piel endurecida[17]. La apariencia de livedo se refiere a una característica clínica donde el eritema reticular se extiende sobre un área amplia[17]; se define como un patrón reticular o moteado de la piel, violáceo, rojo o azul persistente, no reversible con calor, en el tronco, los brazos o las piernas, que consiste en una lesión regular no interrumpida, en general como círculos continuos (livedo reticularis regular) o círculos rotos irregulares (livedo racemosa), este último relacionado con enfermedades como síndrome antifosfolípido, vasculitis y escleroderma, entre otros[20]. Las ulceraciones cutáneas en las extremidades inferiores tienen un curso más prolongado y están relacionadas con la neuropatía[17]. La afectación neurológica se ha reportado en la literatura como neuropatía periférica, defectos focales, hemiplejía, pérdida visual, mononeuritis múltiple, parálisis del nervio renal, psicosis orgánica y convulsiones[18,21]. El compromiso gastrointestinal grave se asoció con un mayor riesgo de recaída y puede manifestarse como infarto o angina mesentérica, perforación, peritonitis, entre otros[22]. Las manifestaciones oculares pueden ocurrir como primera manifestación en un 10-20% de los pacientes; dentro del ojo hay oclusión de la rama y la arteria retiniana central, retinopatía isquémica, pérdida visual monocular transitoria, proptosis, defecto del campo visual bitemporal, neuropatías ópticas isquémicas anteriores o posteriores y es importante reconocer que el proceso vasculítico no se limita a la circulación retiniana: puede presentarse también como conjuntivitis, escleritis, uveítis o neuritis[23]; cuando se afecta el suministro vascular del nervio óptico (principalmente los vasos coroidales y las arterias ciliares posteriores) puede causar edema papilar y papilitis, que, a su vez, puede progresar a atrofia del nervio óptico[23,24], como en el caso de nuestra paciente.
El diagnóstico es predominantemente clínico, aunque la biopsia de tejido y la arteriografía son herramientas importantes en la mayoría de los pacientes. La presencia de angiografía positiva en el momento del diagnóstico es más frecuente en los adultos que en los niños (81% vs. 40%)[25] y muchas veces, como en este caso, los hallazgos en la biopsia y las imágenes se hacen durante el curso y el diagnóstico es puramente presuntivo.
El tratamiento requiere corticoides inicialmente en dosis altas, ya sea por vía oral o intravenosa, que luego se reduce gradualmente durante un período determinado por la respuesta al tratamiento y la gravedad de las manifestaciones clínicas, pero no hay ensayos clínicos que guíen la terapia. Para el tratamiento de inducción a la remisión se utilizan altas dosis de corticoides y CYC entre los primeros 3 a 6 meses; cuando se logra la remisión, la terapia principal de mantenimiento incluye glucocorticoides en dosis bajas (0,2 a 0,4mg/kg/día) y AZA por vía oral en una dosis de 1 a 2 mg/kg/día durante 12 a 18 meses adicionales. También puede usarse metotrexato o leflunomida[13]. Se han descrito algunos casos refractarios manejados con plasmaféresis y otros agentes biológicos, como rituximab, tofacitinib, infliximab y tocilizumab[14,15].
Las complicaciones del tratamiento a corto plazo, como las infecciones y los efectos secundarios inducidos por corticoi-des, continúan siendo una preocupación en los niños con PAN que reciben terapia estándar[6]; además, las complicaciones tardías asociadas con la terapia con CYC incluyen, con mayor frecuencia, alopecia, toxicidad hematológica y, en menor número, toxicidad urológica, malignidad e infertilidad[26] han sido reportadas en cohortes de niños con otras vasculitis[27]. La CH es una manifestación común de urotoxicidad inducida por CYC en enfermedades reumáticas; algunas revisiones han indicado que la vía oral de administración de CYC y las dosis acumuladas podrían aumentar el riesgo de CH[28]; no obstante, aún no está claro si existe una dosis de corte que pueda predecir la aparición de CH[29]. La gravedad de la CH puede variar desde una leve hematuria autolimitada hasta una presentación potencialmente mortal[30], el tiempo promedio de resolución de la hematuria es de 7-56 días, los hallazgos patológicos en las fases agudas y subagudas pueden ser edema, atrofia de la mucosa vesical, ulceración y hemorragia, que se extienden por la pared vesical; en casos severos necrosis y cronicidad, se acompaña, además, de cambios fibróticos del estroma.
En las vasculitis sistémicas, si el sistema genitourinario está involucrado, afecta de manera preferencial a los riñones y los testículos; el compromiso de la vejiga urinaria se ha descrito solo en contados informes de casos y ha surgido como un hallazgo incidental tardío en el curso de la enfermedad[31]. La presentación clínica en general es de síntomas bajos irritativos, los hallazgos radiológicos y en cistoscopia también son inespecíficos, y el diagnóstico se realiza mediante el examen histológico de las biopsias de vejiga, que también sirven para excluir otros diagnósticos diferenciales, especialmente la malignidad[32,33].
Este caso clínico es particularmente llamativo porque logra establecer la presentación novedosa e inusual de la poliarteritis no informada previamente en niños relacionada con actividad de la enfermedad, que en nuestra paciente se vio desencadenada por el curso insidioso de la vasculitis y la falta de adherencia al manejo.
Conclusión
En pacientes con síntomas constitucionales, síndrome febril prolongado y compromiso orgánico, se deben incluir las vasculitis sistémicas en el diagnóstico diferencial. El tratamiento regular es fundamental para la evolución favorable en todas las enfermedades. En esta paciente, una enfermedad severa desde el principio y el tratamiento irregular fueron determinantes de la aparición de una complicación que, por lo conocido hasta el momento, no está descrita en pacientes pediátricos. Requirió igualmente un tratamiento intensivo que incluyó rituximab, medicamento que ha mejorado el pronóstico de pacientes con formas severas de vasculitis.

