










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista EIA
Print version ISSN 1794-1237On-line version ISSN 2463-0950
Rev.EIA.Esc.Ing.Antioq no.15 Envigado July/Dec. 2011
CNN-PROMOTER, NEW CONSENSUS PROMOTER PREDICTION PROGRAM BASED ON NEURAL NETWORKS
CNN-PROMOTER, NUEVO PROGRAMA PARA LA PREDICCIÓN DE PROMOTORES BASADO EN REDES NEURONALES
CNN-PROMOTER, NOVO PROGRAMA PARA A PREDIÇÃO DE PROMOTORES BASEADO EM REDES NEURONAIS
Óscar Bedoya*, Santiago Bustamante**
* Ingeniero de Sistemas y Magíster en Ingeniería de Sistemas, Universidad del Valle. Docente, Escuela de Ingeniería de Sistemas y Computación. Universidad del Valle. Cali, Colombia. oscar.bedoya@correounivalle.edu.co
** Ingeniero de Sistemas, Universidad del Valle. Auxiliar de Investigación, Escuela de Ingeniería de Sistemas y Computación, Universidad del Valle. Cali, Colombia. sbustam@univalle.edu.co
Artículo recibido 24-II-2011. Aprobado 23-VI-2011
Discusión abierta hasta diciembre de 2011
ABSTRACT
A new promoter prediction program called CNN-Promoter is presented. CNN-Promoter allows DNA sequences to be submitted and predicts them as promoter or non-promoter. Several methods have been developed to predict the promoter regions of genomes in eukaryotic organisms including algorithms based on Markov's models, decision trees, and statistical methods. Although there are plenty of programs proposed, there is still a need to improve the sensitivity and specificity values. In this paper, a new program is proposed; it is based on the consensus strategy of using experts to make a better prediction. The consensus strategy is developed by using neural networks. During the training process, the sensitivity and specificity were 100 % and during the test process the model reaches a sensitivity of 74.5 % and a specificity of 82.7 %.
KEY WORDS: Promoter prediction; neural networks; consensus strategy.
RESUMEN
En este artículo se presenta un programa nuevo para la predicción de promotores llamado CNN-Promoter, que toma como entrada secuencias de ADN y las clasifica como promotor o no promotor. Se han desarrollado diversos métodos para predecir las regiones promotoras en organismos eucariotas, muchos de los cuales se basan en modelos de Markov, árboles de decisión y métodos estadísticos. A pesar de la variedad de programas existentes para la predicción de promotores, se necesita aún mejorar los valores de sensibilidad y especificidad. Se propone un nuevo programa que se basa en la estrategia de mezcla de expertos usando redes neuronales. Los resultados obtenidos en las pruebas alcanzan valores de sensibilidad y especificidad de 100 % en el entrenamiento y de 74,5 % de sensibilidad y 82,7 % de especificidad en los conjuntos de validación y prueba.
PALABRAS CLAVE: Predicción de promotores; redes neuronales; estrategia de consenso.
RESUMO
Neste artigo a presenta-se um novo programa para a predição de promotores chamado CNN-Promoter, que toma como entrada sequências de DNA e as classifica como promotor ou não promotor. Desenvolveramse diversos métodos para predizer as regiões promotoras em organismos eucariotas, muitos dos quais se baseiam em modelos de Markov, árvores de decisão e métodos estatísticos. Apesar da variedade de programas existentes para a predição de promotores, precisa-se ainda melhorar os valores de sensibilidade e especificidade. Propõe-se um novo programa que se baseia na estratégia de mistura de experientes usando redes neuronais. Os resultados obtidos nas provas atingem valores de sensibilidade e especificidade de 100 % no treinamento e de 74,5 % de sensibilidade e 82,7 % de especificidade nos conjuntos de validação e prova.
PALAVRAS-CÓDIGO: Predição de promotores; redes neuronais; estratégia de consenso.
1. INTRODUCTION
1.1 Promoter prediction
Gene prediction is a major task in bioinformatics and it can be defined as the problem that takes an uncharacterized DNA sequence as input and identifies the signals frequently observed in genes. Although the structure of a gene is already known, most of the solutions to the problem are supported in determining the elements usually presented in a gene, which are promoter, exons, introns, poly A tail, and transcription start site (TSS). Most of the efforts are focused on trying to design particular models for each of those elements of a gene. The promoter is biologically one of the most important elements of the gene. It is the region upstream of a gene that contains the necessary information for the activation of the gene that it controls (Smale and Kadonaga, 2003).
The promoter region is typically divided into three parts: (1) the core promoter, which is the region typically located ~35 bp upstream of the TSS; (2) the proximal promoter, which is a region containing several regulatory elements and is located a few hundred base pairs upstream of the TSS; and (3) the distal promoter, a region that contains additional regulatory elements called enhancers and silencers. The distal promoter is usually located thousands of base pairs upstream of the TSS. The current available algorithms for promoter prediction do not satisfy the sensitivity and specificity values that biologists would like to obtain. Most of these methods are based on searching motifs in a DNA sequence to decide whether it is a promoter or not (Gordon et al., 2006). The search is usually made by using position weightmatrices and Markov models (Pedersen et al., 1998; Ohler et al., 1999; Liu and States, 2002; Luo, Yang and Liu, 2006; Premalatha, Aravindan and Kannan, 2009; Rymczak and Unold, 2009). Besides statistical strategies, artificial intelligence has also been applied. Particularly, artificial neural networks have shown acceptable sensitive values, but specificity has been affected because of the high false positives rate (Knudsen, 1999; Burden, Lin and Zhang, 2005; Abeel et al., 2008; Zhang, 2009). Neural network has been applied in some other problems in bioinformatics. The motivation behind selecting this technique is related to its capability of identifying hidden patterns in a huge amount of sequences in biological databases. DNA sequences of promoter regions from the same organism present several variations that might be identified using neural networks.
Promoter prediction is particularly difficult in the case of eukaryotic organisms because regulatory regions, such as core promoters and transcription start site, represent just a small percentage of the DNA sequence. Prediction and characterization of regulatory regions is still a challenging problem. There is another issue related to promoter prediction: the existing methods might not coincide with the output for the same uncharacterized DNA sequence as input, which means there is no consensus decision in current predictors.
In this work, a new promoter prediction program is proposed. The program is based on neuronal networks to raise the specificity values maintaining the high sensitivity of the existing models. The strategy used in the proposed program consists of making decisions based on the mixture of three experts. Each expert is a neural network built for well-known consensus sequences such as TATA-box, GC-box, and CAAT-box. Each of these neural networks goes over the sequence identifying a specific box and leaving a mark of "1" indicating the box was found, and "0" otherwise. Then, a major neural network takes the marks left by those experts and tries to make a global prediction.
1.2 Consensus strategy to classify biological sequences
A novel strategy that arises as a hopeful solution to the problem of lack of consensus on classification is the mixture of experts. The strategy has been applied in some other fields of bioinformatics, such as gene prediction (Allen, Pertea and Salzberg, 2004) and secondary structure prediction (Barlow, 1995; De Haan and Leunissen, 2005; Mazo and Bedoya, 2010). The mixture of experts allows running some of the best algorithms for a specific problem and tries to make a decision that integrates the output of the individual methods. The strategy is based on the hypothesis that the consensus decision, the one taken integrating a given number of algorithms, should be better than the individual methods. The consensus decision can be as easy as the majority wins criteria; the decision taken by most of the n given experts is the consensus decision. However, a consensus decision can be taken based on a more complex model, e.g. a decision tree or a neural network.
A decision tree follows a tree structure to make the representation of a given training set. At each node of the tree there is a question that includes one or more attributes in the training data. Each node has one or more branches, each one corresponding to a possible outcome of the question in the node. At the bottom of the tree there are leaf nodes, which assign the classification labels. Decision trees can be used as a mixture of expert strategy in which the question in each node combines the prediction programs. In Allen, Pertea and Salzberg (2004) a decision tree is constructed to predict genes combining the output of three gene predictors: Gene Mark (Lukashin and Bordovsky, 1998), GlimmerM (Pertea and Salzberg, 2002), and Gen Scan (Burge and Karlin, 1997). The consensus model outperforms even the best individual method.
A neural network can also be used as a mixture of expert strategy. In De Haan and Leunissen (2005) a neural network is constructed as a mixture of expert model. The neural network proposed integrates ten secondary structure prediction programs. The model obtained also allows comparing the individual methods by analyzing the weights assigned in the neural network.
In this paper, a new program for promoter prediction is proposed. The program is called CNN-Promoter (CNN stands for Consensus Neural Network) and focuses on combining experts to improve the prediction accuracy. The three experts to be mixed by the neural network are well-known consensus sequences such as TATA-box, GC-box, and CAAT-box.
1.3 Promoter prediction using neural networks
In Kalate, Tambe and Kulkarni (2003) artificial neural networks are used as a tool for predicting mycobacterial promoter sequences and determining structurally/functionally important sub-regions therein. A multi-layered feed-forward neural network of 284 input neurons, one hidden neuron, and one output unit was trained using the error-back-propagation (EBP) algorithm. The network was tested on mycobacterial promoter sequences. According to Kalate, Tambe and Kulkarni (2003), the strategy detects 97 % of the promoters in a test set with mycobacterial promoters and random sequences.
In Zhang, Kuo and Brunkhors (2006) a feed forward neural network is trained to learn E. coli promoters. Coding areas of genes were taken as negative samples. According to Zhang, Kuo and Brunkhors (2006), the network can extract more effectively the statistical characteristics of promoters. Another result demonstrated was that the number of hidden layers seems to have no significant effect on E. coli promoter prediction precision. A CODE-4 orthogonal codification was used as inputs in the neural network. In this codification, each nucleotide in the input layer is represented as four values, i.e., A= 1000, C=0100, G=0010, T=0001. The neural network presents 120 units in the input layer, 80 neurons in the hidden layer, and one node in the output layer. The neural network identified at least 50 % of the promoters in test sets.
Another work that uses neural networks for promoter recognition is Frias, Vidal and Cascardo (2004). It focuses in the genome of the fungus Crinipellis perniciosa. Besides, a new approach for feature extraction, based on local compositional measures, is presented. A feed-forward neural network with just two neurons in the hidden layer was needed. According to Frias, Vidal and Cascardo (2004), 95 % accuracy was obtained.
PromPredictor (Chen and Li, 2005) is a program for recognizing promoter regions. It uses a hybrid neural network approach for predicting promoter regions in large genomic sequences. Prom Predictor is a combination of a novel promoter recognition model, coding theory, feature selection and dimensionality reduction with machine learning algorithm. The method is based on the statistical concept of pentamer distributions in specific functional regions of DNA and selected the most significant pentamer vocabularies from training sequences by an unsupervised learning technique. It is based on a new promoter model with statistical-compositional features and CpG information. According to Chen and Li (2005), sensitivity of 66 % and specificity of 48 % were obtained during testing phase.
Dragon Promoter Finder (DPF) (Bajic et al., 2002) is a program for recognition of vertebrate RNA polymerase II promoters. DPF algorithm identifies TSS positions using five independent promoter recognition models. Each model uses a data window that slides along the DNA sequence. Based on the competition of the models, DPF predicts TSS presence for each data window. According to Bajic et al. (2002), sensitivity of 70 % and specificity of 50 % was obtained.
A consensus strategy is presented in Reese (2001). Two single networks were designed to detect the TATA box and the initiator region. Each neural network was trained independently using TATA-boxes and initiator regions. Combining the two neural networks that were made, time-delay neural networks (TDNN) was used. TDNNs are appropriate for recognizing promoter elements because they can combine multiple features that appear at different relative positions in different sequences. The final input to the TDNN consists of 51 bp, spanning the transcription start site from position -40 to +11, including the TATA-box and the initiator. In this strategy, the outputs of the TATA-box and initiator neural networks are put into a single vector that accumulates the results of the networks. According to Reese (2001), the strategy was able to predict up to 70 % of all promoters in test sets. The organism used in the test set was Drosophila melanogaster.
Most of the works related to design neural networks for promoter prediction have a specific organism used in training, being E. coli, fungus Crinipellis perniciosa, Drosophila melanogaster and myobacterial promoter sequences the most used. Just a few works can be found to be related to recognize human promoters using neural networks. Another issue related to promoter prediction using neural network is that most of the works are based on design networks for specific elements of the promoter, such as TATA-box, initiator region, and transcription start site. There is no neural network method that integrates elements such as CAAT-box, GC-box, and TATA-box in the same consensus model. There is an obvious need for trying a new consensus neural network that combines those elements.
2. MATERIALS AND METHODS
2.1 Selecting and preparing the datasets
Neural networks are based on the idea of training them to learn about the information included in the patterns. The selection of the patterns in the training set is a major decision to make because the generalization capability of the model can be affected. Besides, a test set is also needed, which is a dataset with data not included in the training set that will be used to measure the accuracy of the predicting model.
The datasets used in this work were extracted from the EPD (Eukaryotic Promoter Database). There are a total of 840 promoter sequences of rat and mouse partitioned into 3 datasets which are summarized in table 1. As the non-promoter sequences, a set of short exons and introns were extracted from TIGR (http://www.tigr.org/). The datasets are only composed from promoter sequences DNA type-b, so the results of this paper apply for those kinds of organisms. A total of 80 % of each dataset was used as the training set and 20 % as the test set. In the training set, the training error will be calculated and it indicates the capability of the neural network to learn a set of patterns. Also, when the test set is used, the generalization error will be calculated; it indicates the capability to classify unknown promoters correctly.
In this work, the orthogonal codification CODE-4 is used as the input of the neural networks for the consensus sequences, which is a codification for each nucleotide, C=0001, G=0010, A=0100, and T=1000. As part of the preparation process, every single nucleotide of the datasets was converted to the CODE-4 codification.
2.2 Building neural networks for consensus sequences
As in this paper a mixture of expert strategy is used, it is necessary to build the neural networks for the experts in the consensus sequences. Those are the TATA-box neural network, the GC-box neural network, and the CAAT-box neural network. The topology and some other parameters of those networks are presented as follows:
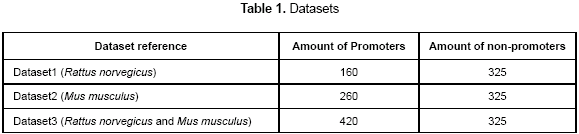
- TATA-box neural network. This neural network presents 28 nodes in the input layer, one neuron in the hidden layer, and one node in the output layer (figure 1). The 28 nodes in the input layer correspond to the codification of the seven nucleotides that usually form the TATA-box. The output of the network will be "1" in case of a 7-nucleotide size subsequence is classified as a positive TATA-box, and "0" otherwise. The neural network uses the logistic function for all of its neurons, and the back-propagation as the learning algorithm. The number of neurons in the hidden layer was modified several times.
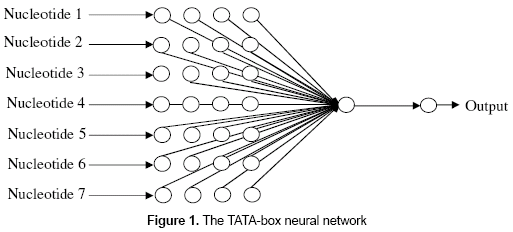
As it was found in Zhang, Kuo and Brunkhors (2006), one neuron was sufficient to let the network learn the patterns in training set. - GC-box neural network. This neural network presents 24 nodes in the input layer, one neuron in the hidden layer, and one node in the output layer (figure 2). The 24 nodes in the input layer correspond to the codification of the six nucleotides that usually form the GC-box. The output of the network will be "1" in case of a 6-nucleotide size subsequence is classified as a positive GC-box and "0" otherwise. The neural network uses the logistic function for all of its neurons, and the back-propagation as the learning algorithm.
- CAAT-box neural network. This neural network is similar to the TATA-box neural network. It also presents 28 nodes in the input layer that correspond to the codification of the seven nucleotides that usually form the CAAT-box.
2.3 Preparing the CNN-Promoter neural network input
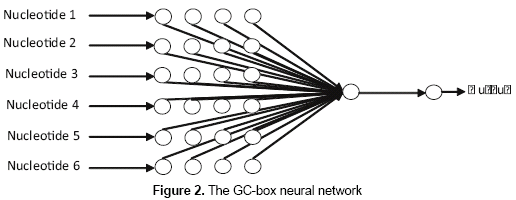
The major difference in this work compared to existing methods is related to how the experts are used. The input for the CNN-Promoter neural network is the marks left by the individual experts, those are, the 1's and 0's left as the outputs of the individual neural networks. Given a DNA sequence, as shown in table 2, there are three rows formed by the outputs of the consensus neural networks. Each row of values or marks is calculated by passing through each consensus neural network over the whole DNA sequence.

The basic idea of the CNN-Promoter neural network is shown in figure 3. The outputs of the three individual experts are calculated from the DNA sequence and taken as the input of the CNN-Promoter.
As in the training set the correct output for each sequence is known, a neural network can be trained to learn how to classify promoter and nonpromoter sequences, taking the marks left by the experts as input. Besides, the relative location of the TATA, GC, and CAAT boxes is included in the model because the positions of the neurons are considered during the classification process.
2.4 Obtaining the CNN-Promoter neural network
A neural network was built for the CNNPromoter program. The network was composed of three layers (input, hidden, and output). The input layer has 732 units. Three input units are necessary for each nucleotide; those three values correspond to the output of the individual experts. The TATA-box, GC-box, and CAAT-box are located from -219 bp to 10 bp from the transcription start site (TSS), which is a total of 230 nucleotides. The window size for the neural network analysis will be 244 nucleotides which is a sufficient length to find the consensus boxes. The 732 units in the layer correspond to the size of the window, which is 244 nucleotides, having three values for each nucleotide. The hidden layer is composed of one unit and the output layer has one neuron; being "1" a promoter and "0" a nonpromoter. Figure 4 shows the configuration of the neural network.
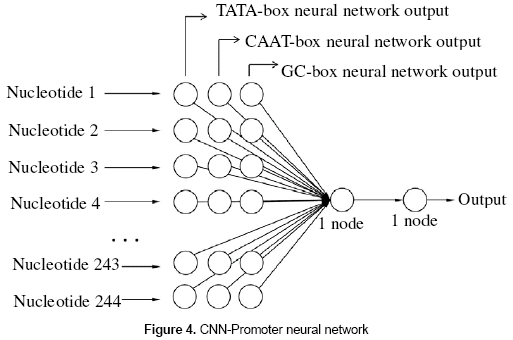
The number of iterations in the learning process was optimized to 200 to save time without losing the learning performance of the network. A logistic function was used as the activation function. In the training process, the whole promoters and non-promoters in the training set were presented to the neural network.
The CNN-Promoter neural network can be used as a classifier of promoters just by feeding the inputs with the uncharacterized DNA sequence. Although, currently there are many promoter predictors, none of them uses the mixture of experts as in this paper. The analysis of marks left by experts is a novel strategy to the promoter prediction problem.
2.5 The CNN-Promoter program
Once the neural network is constructed, a new strategy to classify promoters is obtained. The CNN-Promoter neural network was implemented as a program that allows users to submit DNA sequences and classify them by using the consensus strategy proposed in this paper. The source code of the program is available for academic purposes from the authors upon request.
2.6 Evaluating predictions
The measures used in this paper to evaluate the networks and compare them with some existing strategies are sensitivity (Sn) and specificity (Sp). Sensitivity is the proportion of TP (true positives) predictions out of the total number of actual positives and specificity is the proportion of TN (true negatives) which is correctly identified. Those measures are formally defined as follows:
Sensitivity, Sn=TP/(TP+FN)
Specificity, Sp=TN/(TN+FP)
where TP corresponds to the true positives, FN stands for false negatives and FP corresponds to the false positives. Besides sensitivity and specificity, some other precision measures are incorporated in this study to make a more exhaustive evaluation of the models. The other measures considered are: accuracy, precision, F-measure, and root-mean-square error (RMSE). They are defined as follows:
Accuracy = (TP + TN)/(TP+FP+FN+TN)
Precision = TP/(TP+FP)
F-measure = (2xPrecisionxSensitivity)/(Precision+Sensitivity)
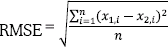 , where x1 is an n-size vector of actual classes and x2 is the vector of predicted classes.
, where x1 is an n-size vector of actual classes and x2 is the vector of predicted classes.
3. RESULTS
A first experiment was related to calculate the capability of the CNN-promoter neural network to learn the Rattus norvegicus (rat) dataset 1. A total of 128 promoters and 260 non-promoters were used as the training set. Sensitivity, specificity, accuracy, precision, F-measure, and RMSE values are shown in table 2. Sensitivity of 100 % and specificity of 100 % were obtained as the training accuracy, which means the topology used is appropriate for promoter prediction. By using the weights in the network calculated previously, a test set was presented to the network. The precision measures were calculated during the test and are also shown in table 3. As can be observed, unseen sequences reduce the accuracy of the model, but they are still acceptable values compared with some of the existing promoter predictors; Promoter Inspector (Scherf, Klingenhoff and Werner, 2000) reports a sensitivity of 48.3 % and a specificity of 43.1 % and First EF (Davuluri, Grosse and Zhang, 2001) reports a sensitivity of 79.3 % and a specificity of 53.5 %.

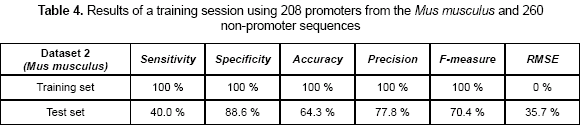
A second experiment was related to calculate the capability of the CNN-promoter neural network to learn the Mus musculus (mouse) dataset 2. In this case, a set of 208 promoters and 260 non-promoters were used as the training set. Sensitivity, specificity, accuracy, precision, F-measure, and RMSE values are shown in table 4. Sensitivity of 100 % and specificity of 100 % were obtained as the training accuracy. By using the weights calculated previously, a test set was presented to the network. The precision measures were calculated during the test and are also shown in table 4.
Another experiment was related to increase the dataset complexity. In this case, both Rattus norvegicus (rat) and Mus musculus (mouse) were used as the positive dataset. A total of 336 promoters and 260 non-promoters were used in the training set. Sensitivity, specificity, accuracy, precision, F-measure, and RMSE values are shown in table 5. Sensitivity of 100 % and specificity of 100 % were obtained as the training accuracy again. By using the weights calculated previously, a test set was presented to the network. The precision measures were calculated during the test and are also shown in table 5.

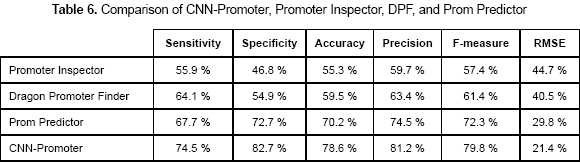
In order to compare the accuracy of CNNPromoter with three of the most representative promoter prediction programs another test was done. The prediction programs used for comparison were Promoter Inspector (Scherf, Klingenhoff and Werner, 2000), Dragon Promoter Finder (DPF) (Bajic et al., 2002), and PromPredictor (Chen and Li, 2005). Table 6 shows the sensitivity and specificity values obtained for the predictors using dataset 3.
4. DISCUSSION
The objective of this project was to obtain a neural network capable of classifying promoters and non-promoters. The CNN-Promoter program uses the strategy of making decisions based on the mixture of three experts. Each expert is also a neural network built for well-known consensus sequences such as TATA-box, GC-box, and CAAT-box. Each of these neural networks goes over the sequence identifying a specific box and leaving a mark of "1" indicating that box was found, and "0" otherwise. Then, a major neural network takes the marks left by those experts and tries to make a global prediction. The analysis of marks left by experts is a novel strategy for the promoter prediction problem and it had never been used.
The network showed perfect accuracy values during the training process which means that topology used was appropriate for the problem of classifying promoters. This accuracy was maintained even when the training set increased its complexity by containing promoters from two different organisms. Table 5 shows sensitivity and specificity values for this particular case. In the test sets, specificity values were at least 75 %, which is a high threshold for the problem of classifying promoters. According to these values, the neural network is capable of identifying a high portion of TP (true positives) out of the total number of promoters predicted, which means that model is just a few times wrong when it classifies a sequence as promoter. Sensitivity values reach the average accuracy of the existing methods; these values indicate that there were promoters in the test set that the model did not identify.
The dataset 3 was the most complex set, but also the more accurate for the neural network. The difference in the training set improves the prediction values. In datasets 1 and 2, sensitivity values are lower than in the dataset 3. This can be explained because the model did have not enough information to generalize using a single organism. When a more complex dataset is used, the model tends to improve the generalization rate, because it has seen more different sequences during the training.
There is a major achievement in this project; the CNN-Promoter is capable of learning guaranteeing 100 % accuracy. It means that biologists could train the network with a particular dataset of their interest and use it, being absolutely sure that the classification of the program is correct. Besides, by comparing the neural network proposed in this work with some of the most remarkable programs for promoter prediction, the CNN-Promoter reaches high values of specificity. As it is shown in table 6, comparing CNN-Promoter with Promoter Inspector, DPF, and Prom Predictor, gives CNN-Promoter the highest accuracy values for the particular dataset used in that test.
The sensitivity, as shown in the experiments, can be improved by selecting a more complex dataset for the training process. The CNN-Promoter could also be improved by integrating additional experts such as the initiation codon and some other signals related to the promoter composition.
REFERENCES
Abeel, T.; Saeys, Y.; Bonnet, E.; Rouze, P. and Van de Peer, Y. (2008). "Generic eukaryotic core promoter prediction using structural features of DNA". Genome Research, vol. 18, No. 2 (February), pp. 310-323. [ Links ]
Allen, J. E.; Pertea, M. and Salzberg, S. L. (2004). "Computational gene prediction using multiple sources of evidence". Genome Research, vol. 14, No. 1 (January), pp. 142-148. [ Links ]
Bajic, V; Seah, S.; Chong, A; Zhang, G; Koh, J. L. Y. and Brusic, V (2002). "Dragon Promoter Finder: Recognition of vertebrate RNA polymerase II promoters". Bioinformatics, vol. 18, No. 1 (January), pp. 198-199. [ Links ]
Barlow, T. W. Feed-forward neural networks for secondary structure prediction. (1995). Journal of Molecular Graphics and Modelling, vol. 13, No. 3 (June), pp. 175-183. [ Links ]
Burden, S.; Lin, Y.-X. and Zhang, R. (2005). "Improving promoter prediction for the NNPP2.2 algorithm: A case study using Escherichia coli DNA sequences". Bioinformatics, vol. 21, No. 5 (March), pp. 601-607. [ Links ]
Burge, C. and Karlin, S. (1997). "Prediction of complete gene structures in human genomic DNA". Journal of Molecular Biology, vol. 268, No. 1 (April), pp. 78-84. [ Links ]
Chen, C. B. and Li, T. (2005). "A hybrid neural network system for prediction and recognition of promoter regions in human genome". Journal of Zhejiang University Science B, vol. 6, No. 5 (May), pp. 401-407. [ Links ]
Davuluri, R; Grosse, I. and Zhang, M. (2001). "Computational identification of promoters and first exons in the human genome". Nature Genetics, vol. 29, No. 4 (December), pp. 412-417. [ Links ]
De Haan, Jorn R. and Leunissen, Jack A. M. (2005). "Protein secondary structure prediction: Comparison of ten common prediction algorithms using a neural network". Nato Science Series Sub Series I. Life and Behavioural Sciences, vol. 368, pp. 149-161. [ Links ]
Frias, D.; Vidal, R.; Cascardo, J. C. M. Finding gene promoters in the genome of the fungus Crinipellis perniciosa using feed-forward neural networks. Machine Learning for Signal Processing, 2004. Proceedings of the 2004 14th IEEE Signal Processing Society Workshop. 2004. [ Links ]
Gordon, J. J.; Towsey, M. W.; Hogan, J. M.; Mathews S. A. and Timms, P. (2006). "Improved prediction of bacterial transcription start sites". Bioinformatics, vol. 22, No. 2, pp. 142-148. [ Links ]
Kalate, R. N.; Tambe, S. S. and Kulkarni, B. D. (2003). "Artificial neural networks for prediction of mycobacterial promoter sequences". Computational Biology and Chemistry, vol. 27, No. 6 (December), pp. 555-564. [ Links ]
Knudsen, S. (1999). "Promoter 2.0: For the recognition of Pol II promoter sequences". Bioinformatics, vol. 15, No. 5, pp. 356-361. [ Links ]
Liu, R. and States, D. (2002). Consensus promoter identification in the human genome utilizing expressed gene markers and gene modelling. Genome Research, 12, pp. 462-469. [ Links ]
Lukashin, A. V and Bordovsky, M. (1998). "GeneMark. hmm: New solutions for gene finding". Nucleic Acids Research, vol. 26, No. 4, pp. 1107-1115. [ Links ]
Luo, Q.; Yang, W. and Liu P. (2006). "Promoter recognition based on the interpolated Markov chains optimized via simulated annealing and genetic algorithm". Pattern Recognition Letters, vol. 27, No. 9 (July), pp. 1031-1036. [ Links ]
Mazo, Claudia y Bedoya, Óscar. (2010). "PESPAD: Una nueva herramienta para la predicción de la estructura secundaria de la proteína basada en árboles de decisión". Ingeniería y Competitividad, vol. 12 (diciembre), No. 2, p. 9-22. [ Links ]
Ohler, U.; Harbeck, S.; Niemann, H.; Noth, E. and Reese M. G. (1999). "Interpolated Markov chains for eukaryotic promoter recognition". Bioinformatics, vol. 15, No. 5, pp. 362-369. [ Links ]
Pedersen A.; Baldi P.; Brunak, S. and Chauvin, Y. Characterizacion of prokariotic and eukariotic promoters using hidden Markov models. Proceedings of the Third International Conference on Intelligent Systems for Molecular Biology. (ISMB98), 1998. [ Links ]
Pertea, M. and Salzberg, S. L. (2002). "Computational gene finding in plants". Plant Molecular Biology, vol. 48, No. 1-2, pp. 39-48. [ Links ]
Premalatha, C.; Aravindan, C. and Kannan, K. On improving the performance of promoter prediction classifier for eukaryotes using fuzzy based distribution balanced stratified method. Proceedings of the International Conference on Advance in Computing, Control, and Telecommunication Technologies, 2009. ACT 2009, IEEE, (28-29 December), pp. 364-366. [ Links ]
Reese, M. G. (2001). "Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome". Computer & Chemistry, vol. 26, No. 1 (December), pp. 51-56. [ Links ]
Rymczak, K. and Unold, O. Improved (non)fixed TSS methods for promoter prediction. Proceedings of the International Multiconference on Computer Science and Information Technology, 2009. IMCSIT 2009, IEEE, (12-14 October), pp.105-108. [ Links ]
Scherf, M.; Klingenhoff, A. and Werner, T. (2000). "Highly specific localization of promoter regions in large genomic sequences by Promoter Inspector: A novel context analysis approach". Journal of Molecular Biology, vol. 297, No. 3 (March), pp. 599-606. [ Links ]
Smale, S. and Kadonaga, J. (2003). "The RNA polymerase II core promoter". Annual Review of Biochemistry, vol. 72 (July), pp. 449-479. [ Links ]
Zhang, F.; Kuo, M.D. and Brunkhors, A. "E. coli promoter prediction using feed-forward neural networks". Engineering in Medicine and Biology Society, EMBS '06. 28th Annual International Conference of the IEEE. 2006. [ Links ]
Zhang, Ya-Jing. A novel promoter prediction method inspiring by biological immune principles. Global Congress on Intelligent Systems, 2009. GCIS 2009. Xiamen, China (19-21 May), vol. 1, pp. 569-573. [ Links ]

